MERCURIUS™ Total BRB-seq is a total RNA-seq kit for purified RNA samples, designed for scalable whole-transcriptome profiling when coding and non-coding transcript information is required.
The workflow combines rRNA depletion, early sample barcoding, and multiplexing of up to 96 purified RNA samples in a single tube. By preserving full-length transcript coverage, MERCURIUS™ Total BRB-seq supports applications such as differential expression analysis, regulatory RNA studies, isoform and splice variant analysis, biomarker discovery, toxicogenomics, and richer transcriptomic dataset generation.
Total RNA-seq enables broader transcriptome profiling than poly(A)-selected or 3′ mRNA-seq workflows by capturing both coding and non-coding RNA species. This makes it useful when regulatory RNAs, antisense transcripts, isoforms, splice variants, or broader whole-transcriptome information may contribute to the biological question.
For gene-level expression profiling at the lowest cost per sample, 3′ BRB-seq may be the better fit. For broader transcript coverage, choose Total BRB-seq.
Saturation curves indicating the number of detected genes as a function of sequencing depth in Huh7 cells. MERCURIUS™ Full-Length BRB-seq and Total BRB-seq perform comparably to NEBNext® Ultra™ II mRNA and Total RNA preparations, respectively, demonstrating equivalent sensitivity at a fraction of the library preparation cost and effort. MERCURIUS™ BRB-seq is a 3’ mRNA library preparation technology, hence the lower overall gene detection versus full-length methods.

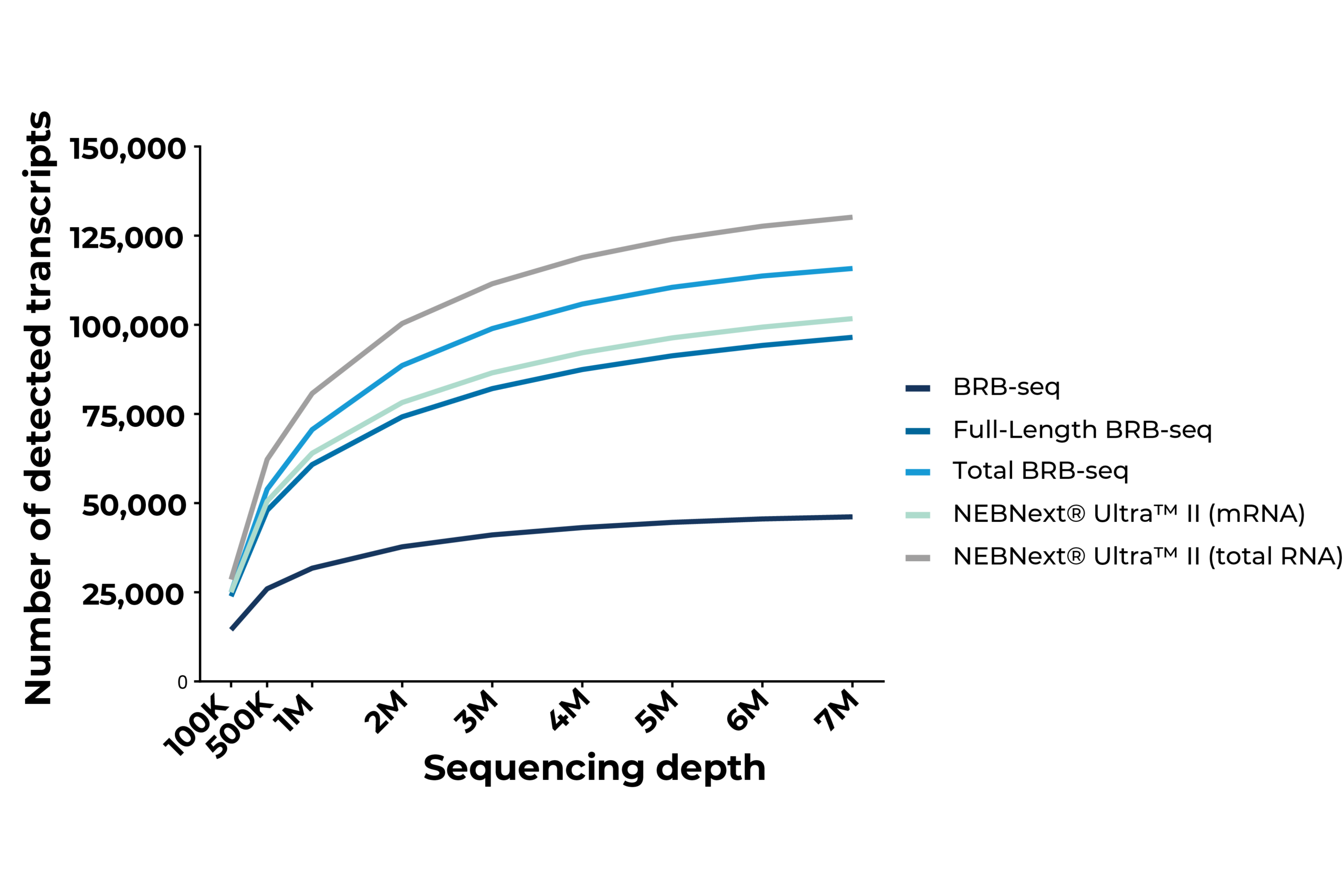
Saturation curves indicating the number of detected transcripts as a function of sequencing depth in Huh7 cells. MERCURIUS™ Full-Length BRB-seq and Total BRB-seq perform comparably to NEBNext® Ultra™ II mRNA and Total RNA preparations, respectively, demonstrating equivalent sensitivity at a fraction of the library preparation cost and effort. MERCURIUS™ BRB-seq specifically detects protein-coding transcripts, hence the lower overall transcript detection compared to full-length methods.
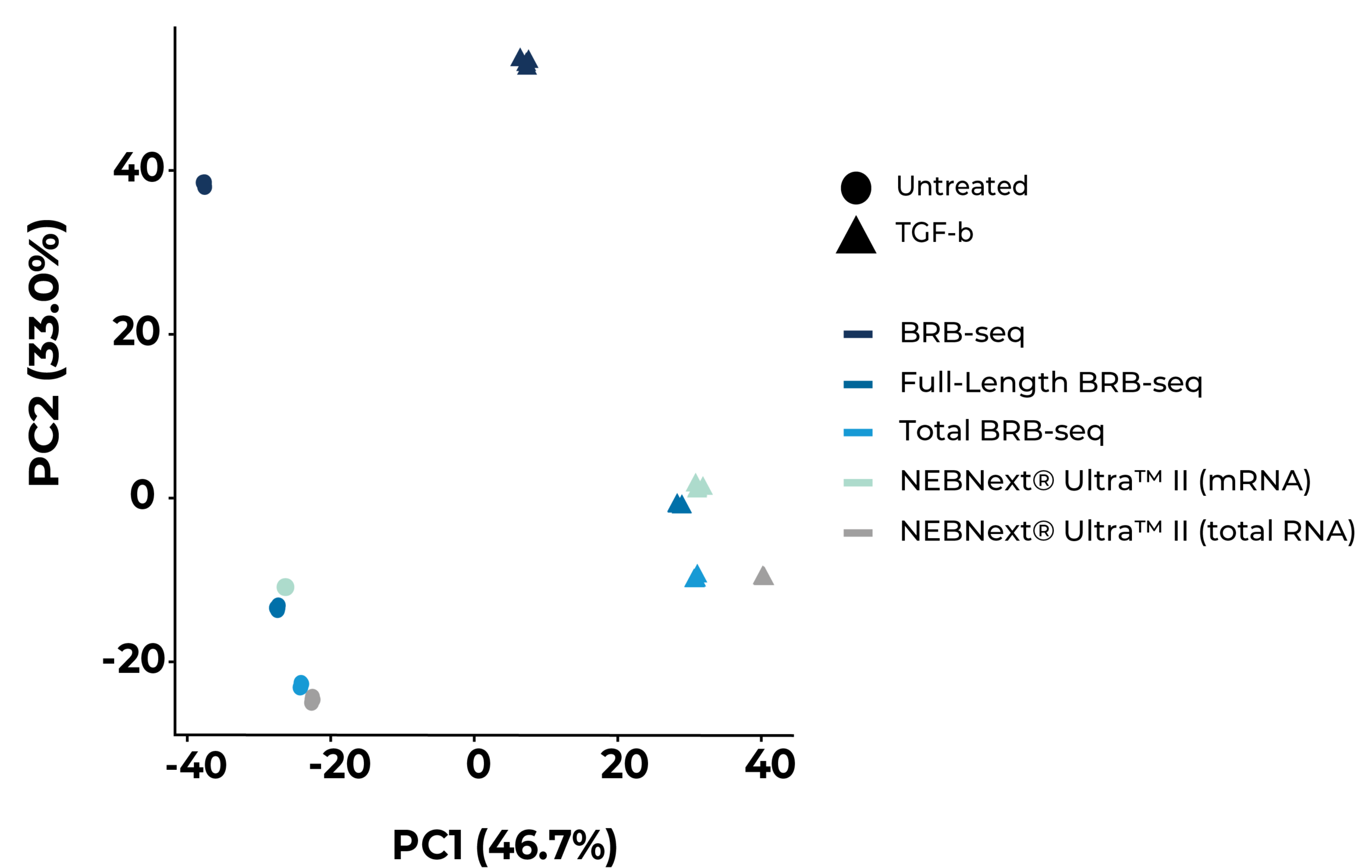
Principal component analysis (PCA) of gene expression profiles generated by the specified library preparation methods across untreated (circles) and TGF-β-treated (triangles) Huh7 samples. Samples cluster primarily by library type along PC2 and by treatment condition along PC1. All methods robustly capture the expression response to treatment. MERCURIUS™ Full-Length BRB-seq and Total BRB-seq cluster closely with their NEBNext® Ultra™ II counterparts, demonstrating equivalent transcriptomic profiles. MERCURIUS™ BRB-seq clusters separately, consistent with its 3′-end capture design.
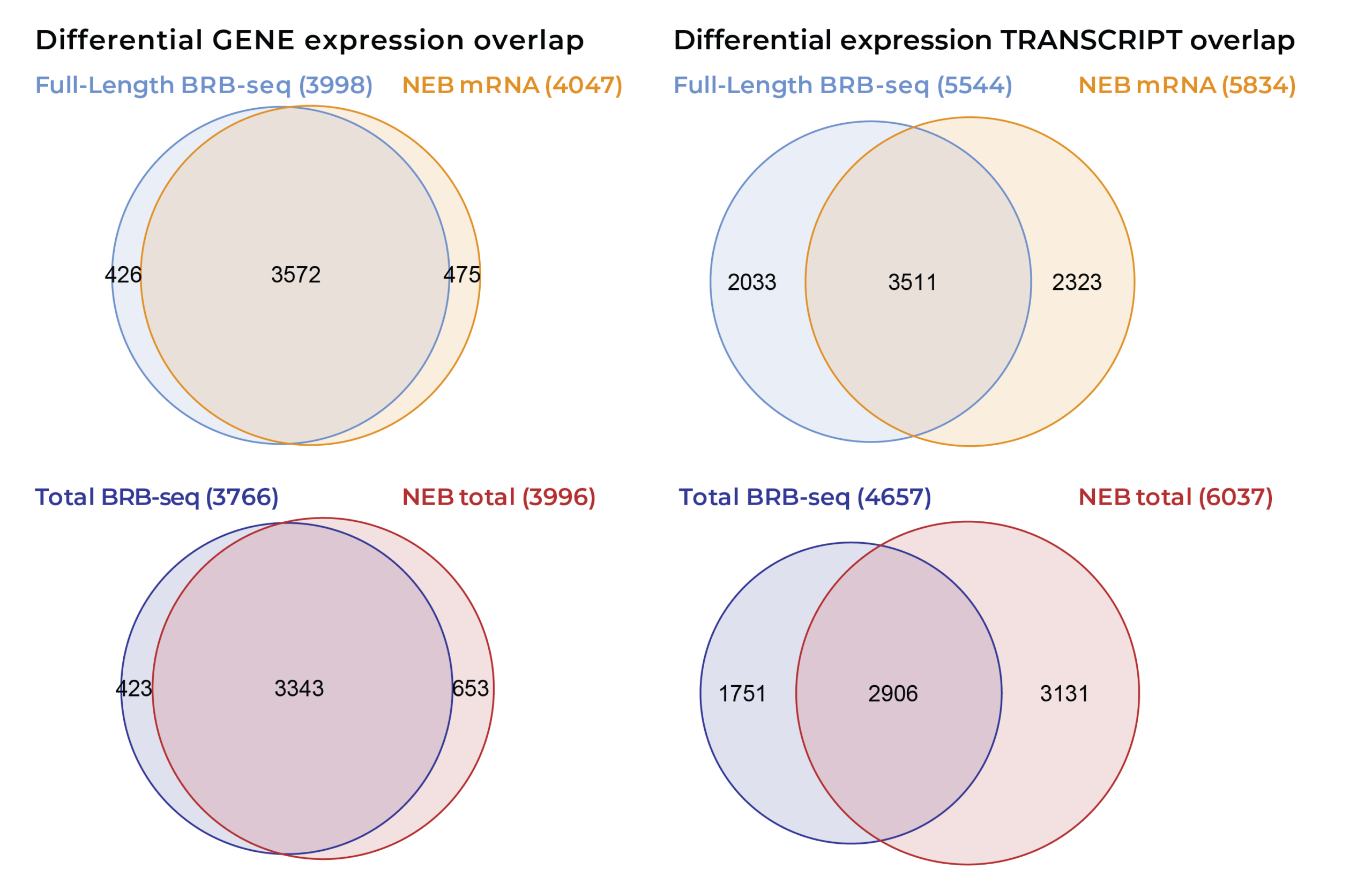
Venn diagrams showing the overlap in differentially expressed (DE) genes (left) and transcripts (right) identified by MERCURIUS™ Full Length BRB-seq and NEBNext® Ultra™ II for mRNA libraries (top) and total RNA libraries (bottom). Numbers indicate DE features unique to each method or shared between both. At the gene level, MERCURIUS™ Full Length BRB-seq and NEBNext® Ultra™ II share over 88% of DE genes for mRNA and 84% for total RNA preparations, demonstrating strong concordance in differential expression calls across both library types. The overlap is smaller but still strong for DE transcripts across technologies.
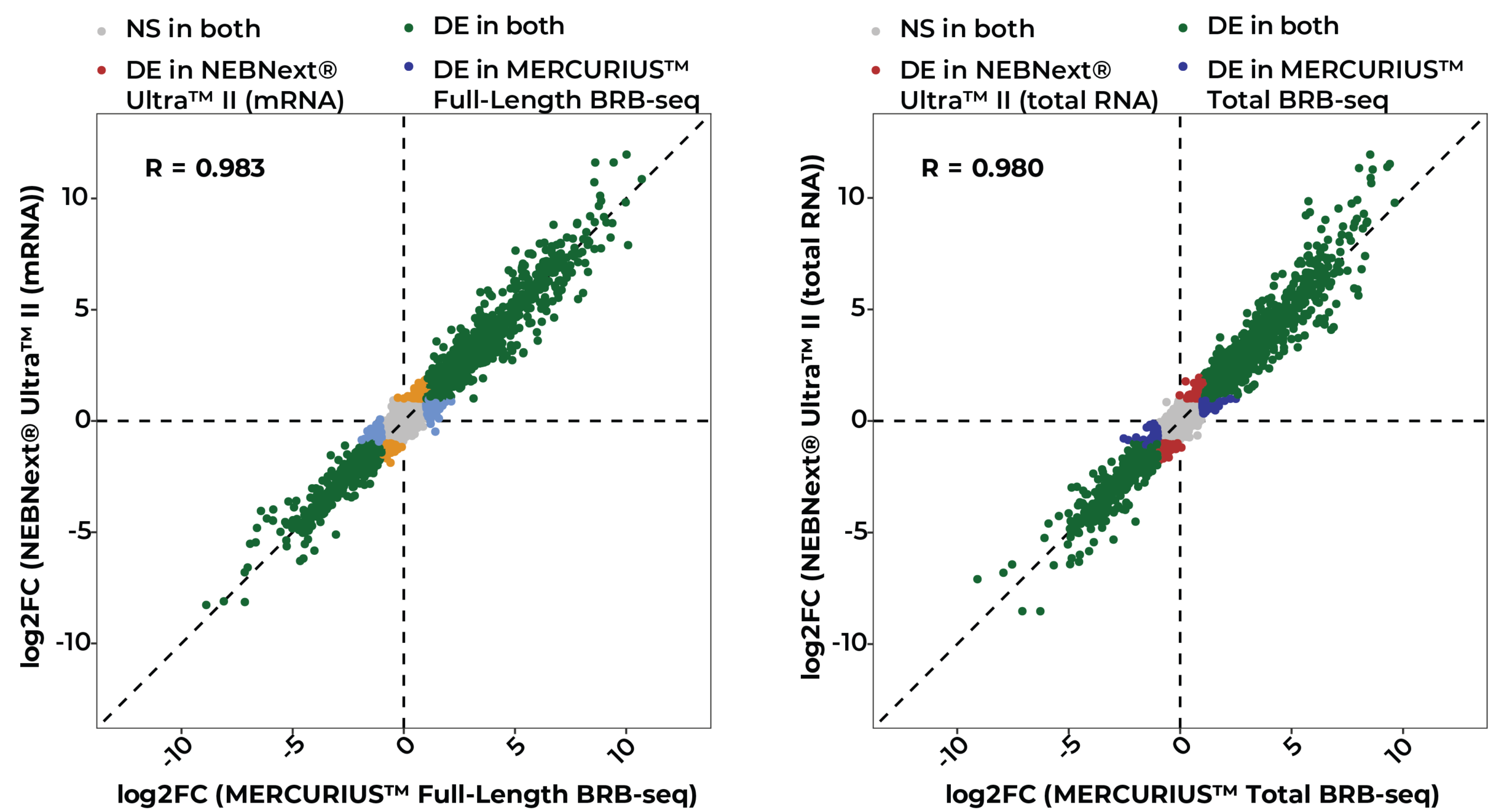
Log2 fold-change correlation between MERCURIUS™ Full-Length BRB-seq and NEBNext® Ultra™ II for mRNA (left, R = 0.983) and total RNA (right, R = 0.980) protocols, across intersecting gene sets of 13,364 and 13,237 genes, respectively. Each point represents a gene, colored by its differential expression (DE) or non-significant (NS) status after treatment of Huh7 cells with human TGF-β versus untreated cells. The dashed diagonal represents perfect correlation. The concordance of DE fold changes across both RNA input types demonstrates that MERCURIUS™ Full-Length and Total BRB-seq reliably recapitulate the results of gold-standard library preparation.
A total RNA-seq kit prepares sequencing libraries from total RNA, enabling broader transcriptome profiling than mRNA-only approaches. Depending on the workflow, total RNA-seq can capture coding and non-coding transcripts after ribosomal RNA depletion.
Each kit includes 4 Unique Dual Index (UDI) pairs, so you can prepare up to 4 indexed libraries (pools) per kit.
The tested input range is 100 pg to 100 ng total RNA per well. For best results (library complexity + uniformity), aim for 10–100 ng per well, and pool at least 8 wells per library.
Because Total BRB-seq uses early multiplexing, it’s important that RNA quantity and quality are consistent across samples (e.g., RIN > 7, 260/230 > 1.5, and ~±10% input uniformity).
Total BRB-seq is designed for total RNA sequencing (not just mRNA), including coding and non-coding transcripts, and includes an rRNA depletion step prior to library prep.
We recommend 10-20 million reads for each sample, which enables the detection of 65,000 transcripts.
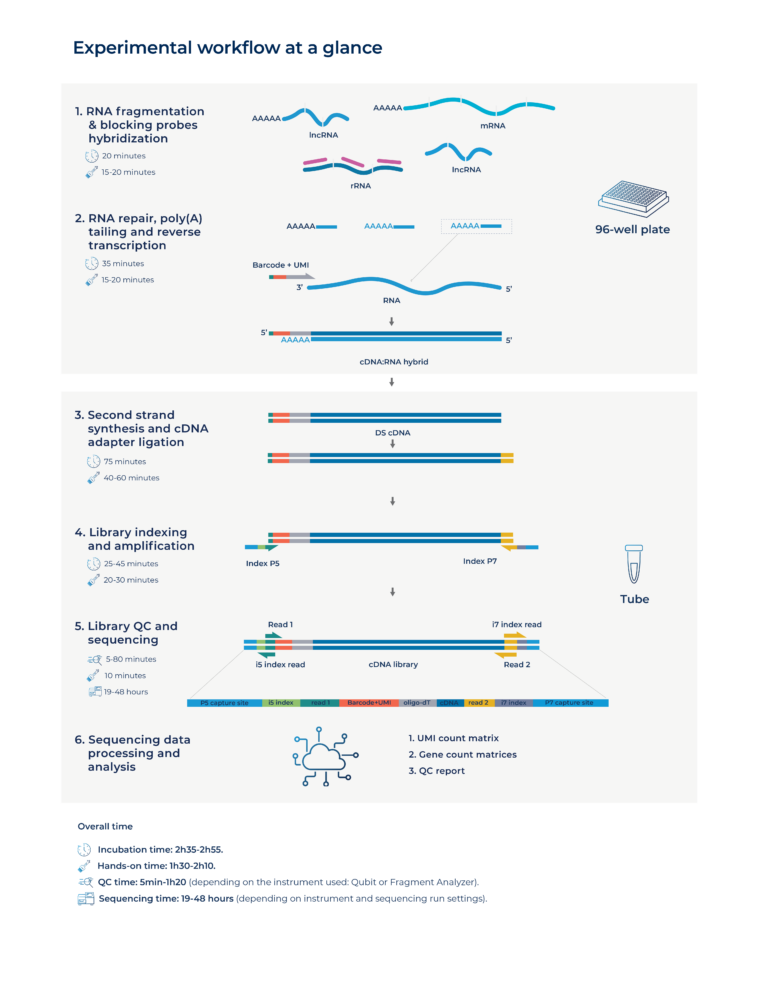
Yes, there is one extra step: after standard index demultiplexing, you demultiplex by sample barcodes to generate per-sample gene/transcript count outputs. The workflow supports producing outputs like UMI count matrices, gene count matrices, and a QC report.
Go to the “Resources” section of this page and click on the button to download the barcode files for the product you are using.
Yes. If you are not ready to run kits internally, Alithea can run the workflow and deliver FASTQ files, gene count matrices, and analysis report files.
* Contact us to inquire about compatibility with other species.
Product
Catalog Number
MERCURIUS™ UDI X-Leap Expansion module
MERCURIUS™ Full-Length Post-Pooling Preparation Module (4 libraries)
Each kit includes 4 UDI pairs. Add the expansion module if you need more unique indexes (total 16 UDI pairs available).
Determining the most suitable transcriptomic technology to drive your large-scale compound screen, clinical study, or to assess a panel of genetic perturbations can be a…
High-throughput’ in sequencing refers to the amount of DNA molecules read at the same time. Technologies are now capable of sequencing many fragments of DNA…
With a growing number of published 3’ mRNA-seq methods now available, researchers have more choices than ever for high-throughput and cost-effective transcriptomic screening. While broadly…