Discover Sample Datasets
Mammals, Plants, Insects, Fish
MERCURIUS™ BRB-seq is a bulk RNA-seq kit for researchers who need cost-effective gene expression profiling across many purified RNA samples. The workflow is designed for academic labs, core facilities, and research teams running studies with larger cohorts, multiple conditions, time points, tissues, species, or perturbations.
By combining early sample barcoding, unique molecular identifiers, and sample pooling, MERCURIUS™ BRB-seq reduces workflow complexity and makes scalable 3′ mRNA-seq practical for differential gene expression analysis, transcriptome profiling, and large experimental designs.
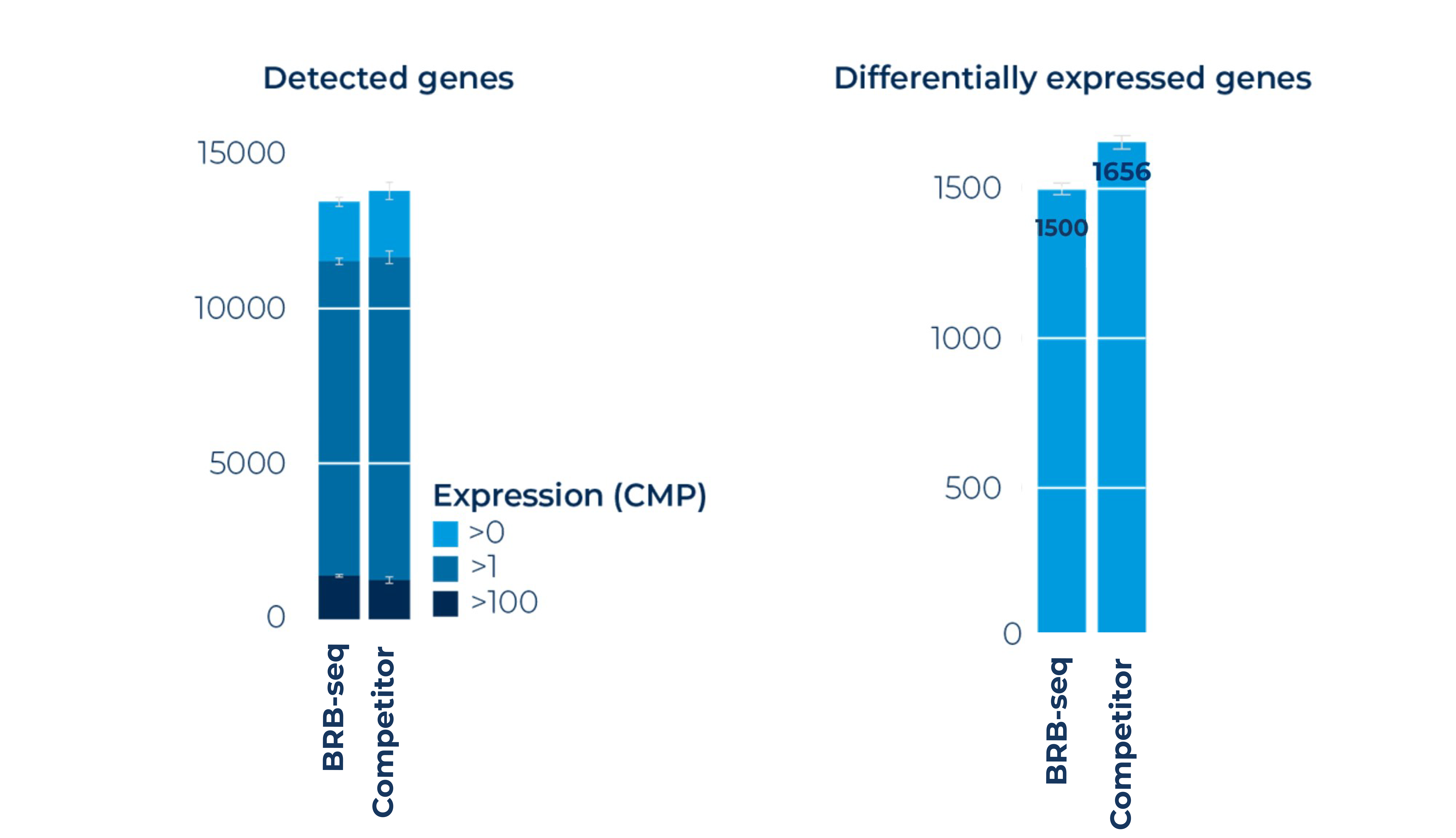
Comparison of gene detection (left) and differential expression (right) between the same samples prepared with MERCURIUS™ BRB-seq or Illumina TruSeq. (Left) Both protocols detect equivalent numbers of genes across all expression thresholds at the same sequencing depth. (Right) Differential expression analysis yields comparable numbers of differentially expressed genes for both technologies. MERCURIUS™ BRB-seq achieves TruSeq-equivalent analytical performance at significantly higher throughput and lower cost per sample.
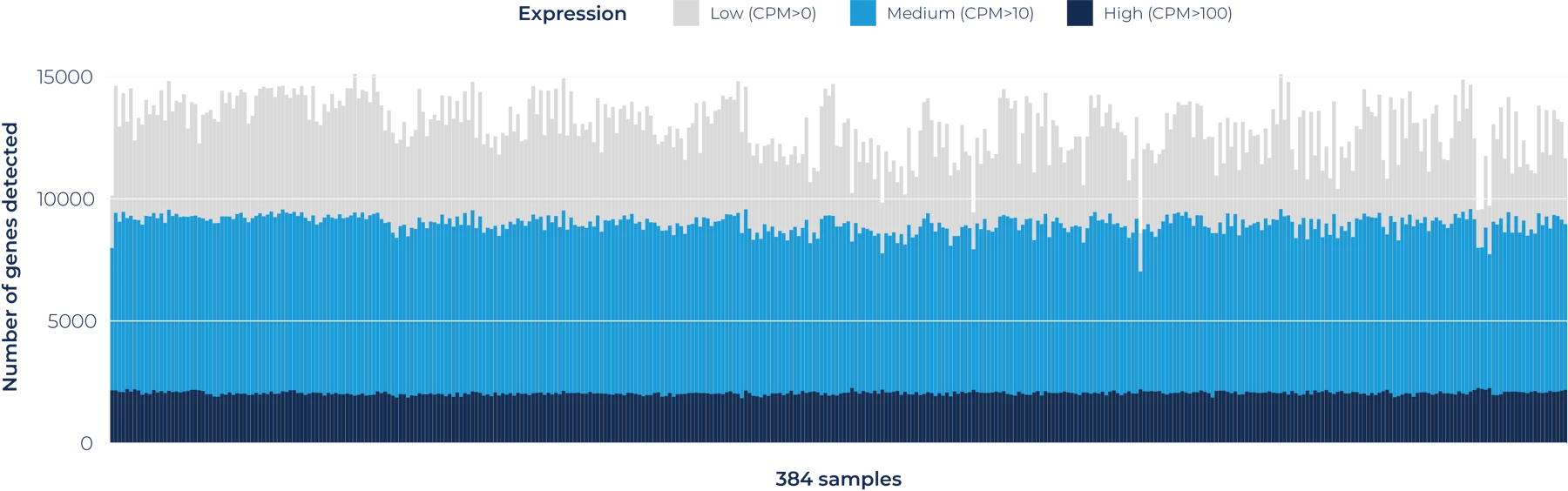
Number of genes detected per sample across a 384-sample BRB-seq run, stratified by expression level. Across all 384 samples, an average of ~13,000 genes is detected when sequenced at a depth of 1.5M reads/sample. The uniformity of gene detection across all samples demonstrates the reproducibility and scalability of MERCURIUS™ BRB-seq for high-throughput transcriptomic profiling.
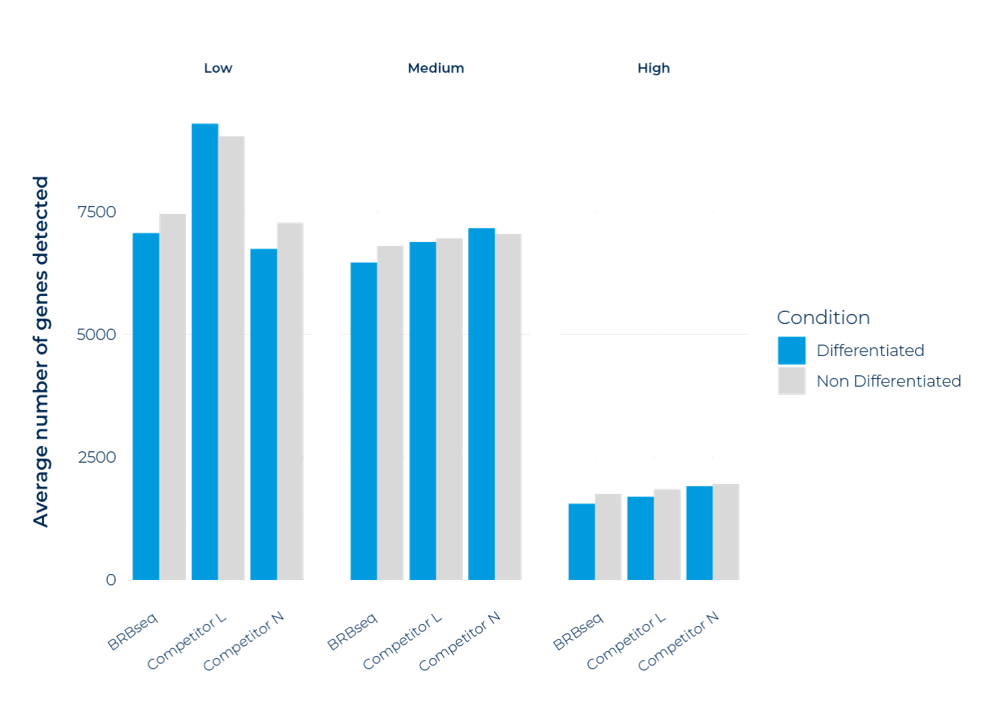
Average number of genes detected per expression category in non-differentiated (ND) and differentiated (D) adipose stromal population cells, comparing MERCURIUS™ BRB-seq against two commercial competitor kits. Genes are stratified into lowly expressed (0 < CPM < 10), mid-expressed (10 < CPM < 100), and highly expressed (CPM > 100) categories. All replicates were prepared from the same RNA sample and sequenced at a uniform depth of 3 million reads per sample. MERCURIUS™ BRB-seq performs comparably to both competitors across all expression levels and cell states.
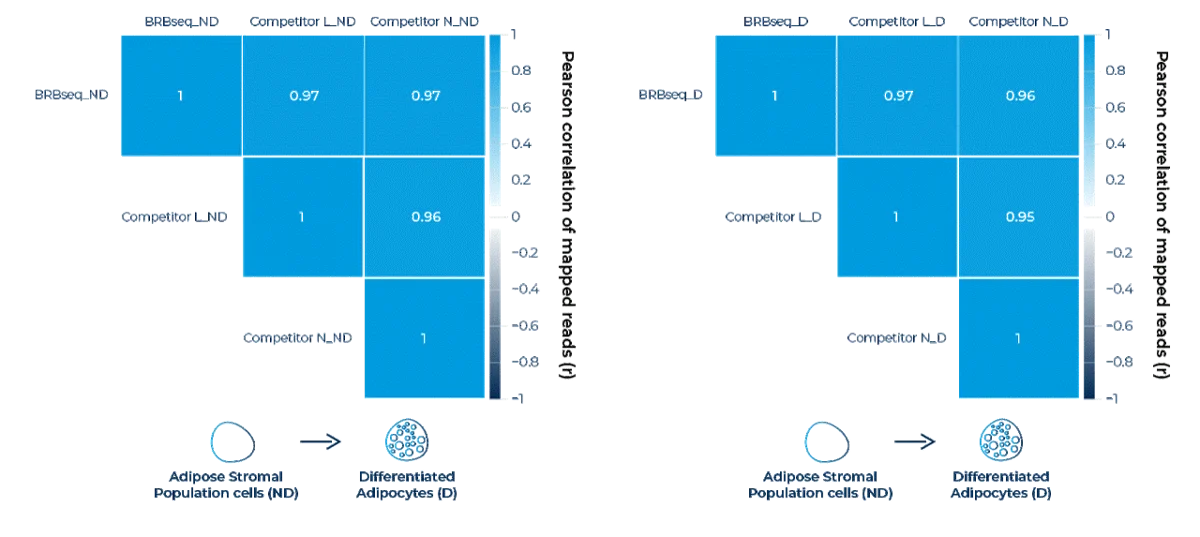
Transcriptome-wide Pearson correlation matrices of mapped reads comparing MERCURIUS™ BRB-seq against Competitor L and Competitor N in non-differentiated adipose stromal Population cells (ND, left) and differentiated adipocytes (D, right). MERCURIUS™ BRB-seq achieves Pearson correlation coefficients of r ≥ 0.97 against both competitors in both cell states, comparable to the inter-competitor correlation (r = 0.95–0.96), demonstrating that MERCURIUS™ BRB-seq produces highly concordant gene expression profiles regardless of biological condition.
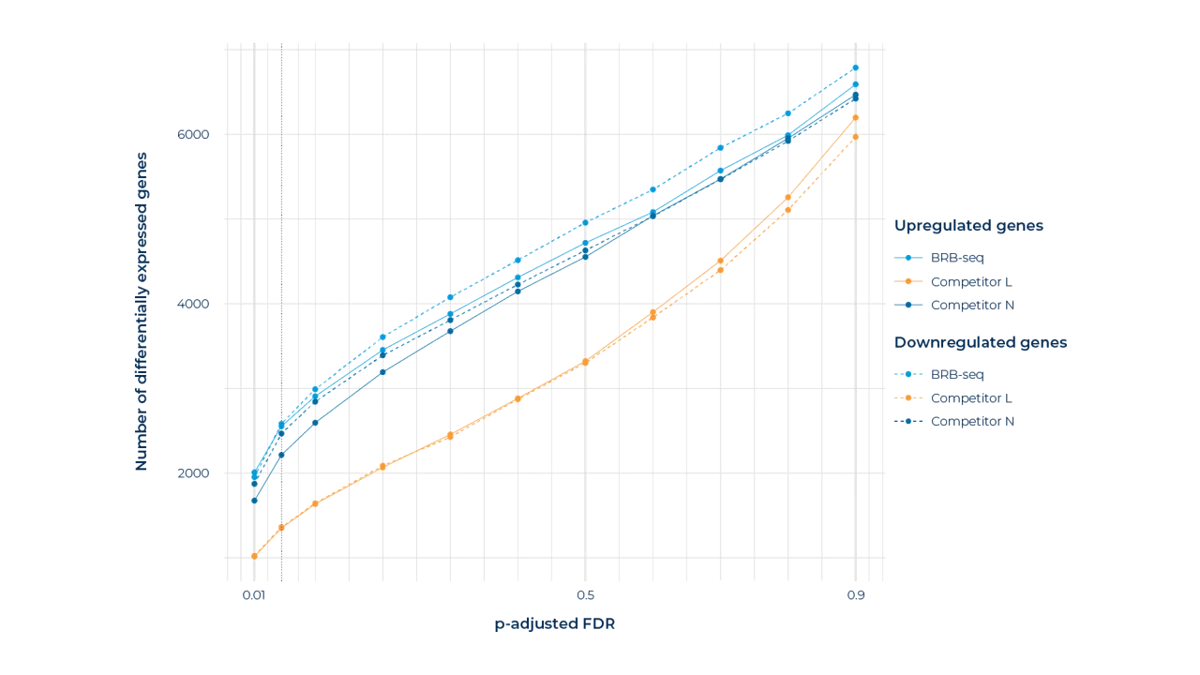
Number of up and downregulated differentially expressed genes (DEGs) detected by MERCURIUS™ BRB-seq, Competitor L, and Competitor N as a function of FDR-adjusted p-value threshold, comparing non-differentiated adipose stromal population cells to differentiated adipocytes at 3M reads/sample on the same initial RNA. MERCURIUS™ BRB-seq consistently identifies more DEGs than both competitors across the full range of significance thresholds, suggesting greater sensitivity for capturing the full transcriptional response to differentiation.
To have access to the deep-sequenced dataset (9.5M reads per sample) contact us.
To have access to the deep-sequenced dataset (8.9M reads per sample) contact us.
To have access to the deep-sequenced dataset (5.7M reads per sample) contact us.
To have access to the deep-sequenced dataset (6.3M reads per sample) contact us.
To have access to the deep-sequenced dataset (9M reads per sample) contact us.
To have access to the deep-sequenced dataset (3M reads per sample) contact us.
To have access to the deep-sequenced dataset (9M reads per sample) contact us.
To have access to the deep-sequenced dataset (3M reads per sample) contact us.
To have access to the deep-sequenced dataset (4M reads per sample) contact us.
To have access to the deep-sequenced dataset (5.3M reads per sample) contact us.
To have access to the deep-sequenced dataset (4.7M reads per sample) contact us.
Each BRB-seq kit contains reagents (including four pairs of Unique Dual Indexing adapters) sufficient for the complete library preparation process for four different BRB-seq pools.
To note, the total number of RNA samples that can be processed with one kit does not exceed the kit specifications; for instance, a 96-sample kit can be used to prepare up to 96 samples distributed across up to four different libraries.
The recommended range of RNA amount for each sample is 50-1000 ng. Normally, the more RNA, the better.
The minimum recommended RIN number is 6 and the A260/230 ratio (Nanodrop) should be in the 1.5-2.2 range.
The only difference between BRB-seq and standard RNA-seq data analysis is the demultiplexing step, which is used to assign sequencing reads to their sample of origin based on the BRB-seq barcode sequence.
For a thorough description of BRB-seq data processing, please refer to the BRB-seq kit data analysis user guide.
One of the key advantages of BRB-seq is that it does not only save reagents and cost in the library preparation stage, but also in the sequencing one.
As opposed to standard RNA-seq, where 20M-30M reads per sample are required, we normally recommend to sequence BRB-seq libraries at a depth of 4M-5M reads per sample, which is normally enough to detect the vast majority of expressed genes.
The barcode set for your kit is conveniently located on the kit label. Please refer to the label for accurate identification.
For optimal compatibility, ensure that you use the appropriate plate format (e.g., for kits designed for 96 reactions, the 96 well-plate format should be used). This ensures accurate and efficient processing of your samples. If you have any further questions or concerns, please contact our support team for assistance by email or using our live chat tool.
* Contact us to inquire about compatibility with other species.
Product
MERCURIUS™ UDI Expansion module
MERCURIUS™ Standard Post-Pooling Preparation Module (4 libraries)
Each kit includes 4 UDI pairs. Add the expansion module if you need more unique indexes (total 16 UDI pairs available).
Determining the most suitable transcriptomic technology to drive your large-scale compound screen, clinical study, or to assess a panel of genetic perturbations can be a…
High-throughput’ in sequencing refers to the amount of DNA molecules read at the same time. Technologies are now capable of sequencing many fragments of DNA…
With a growing number of published 3’ mRNA-seq methods now available, researchers have more choices than ever for high-throughput and cost-effective transcriptomic screening. While broadly…
In this post, we will see briefly how to perform downstream analysis for RNA-seq data.
…