Extraction-free and high-throughput RNA-seq technology designed to provide unbiased and high-content full transcriptome profiling across thousands of samples in parallel at low cost and short turnaround times.
MERCURIUS™ DRUG-seq turns high-throughput transcriptomic profiling into a practical decision tool for large-scale compound discovery programs, next-generation toxicology strategies, and mechanism-driven safety assessment. The result is faster insight, better compound prioritization, and more informed decisions across the discovery and safety pipeline.
MERCURIUS™ DRUG-seq detects compound pathway engagement, distinguishes universal cellular stress responses from compound-specific signatures, and uncovers hidden biology, such as off-target activity, shifts in cell differentiation, and immune activation, challenging to detect with other approaches.
By delivering standardized scoring and robust cross-study comparability, MERCURIUS™ DRUG-seq enables researchers to cluster compounds by mechanism of action, prioritize biological pathways, and track dose- and time-dependent responses for more confident go/no-go decisions.

Quantify pathway engagement, distinguish shared stress programs from compound-specific signatures, and surface unexpected biology such as off-target effects, differentiation shifts, and immune activation early in the pipeline.
Move from cell lysates to sequencing-ready libraries with fewer steps and less hands-on time. By combining direct-from-lysate processing with an improved reverse transcription step, MERCURIUS™ DRUG-seq avoids the template-switching oligos and pre-amplification used in the original protocol, simplifying the workflow while improving mapping and gene detection rates.
Run DRUG-seq in 96-, 384-, or 1,536-well plate formats, depending on the stage and scale of your study. Early barcoding and pooling let you process many samples together from the start of the workflow, making it practical to run transcriptomic studies at the scale needed for discovery, development, and cross-study comparison.
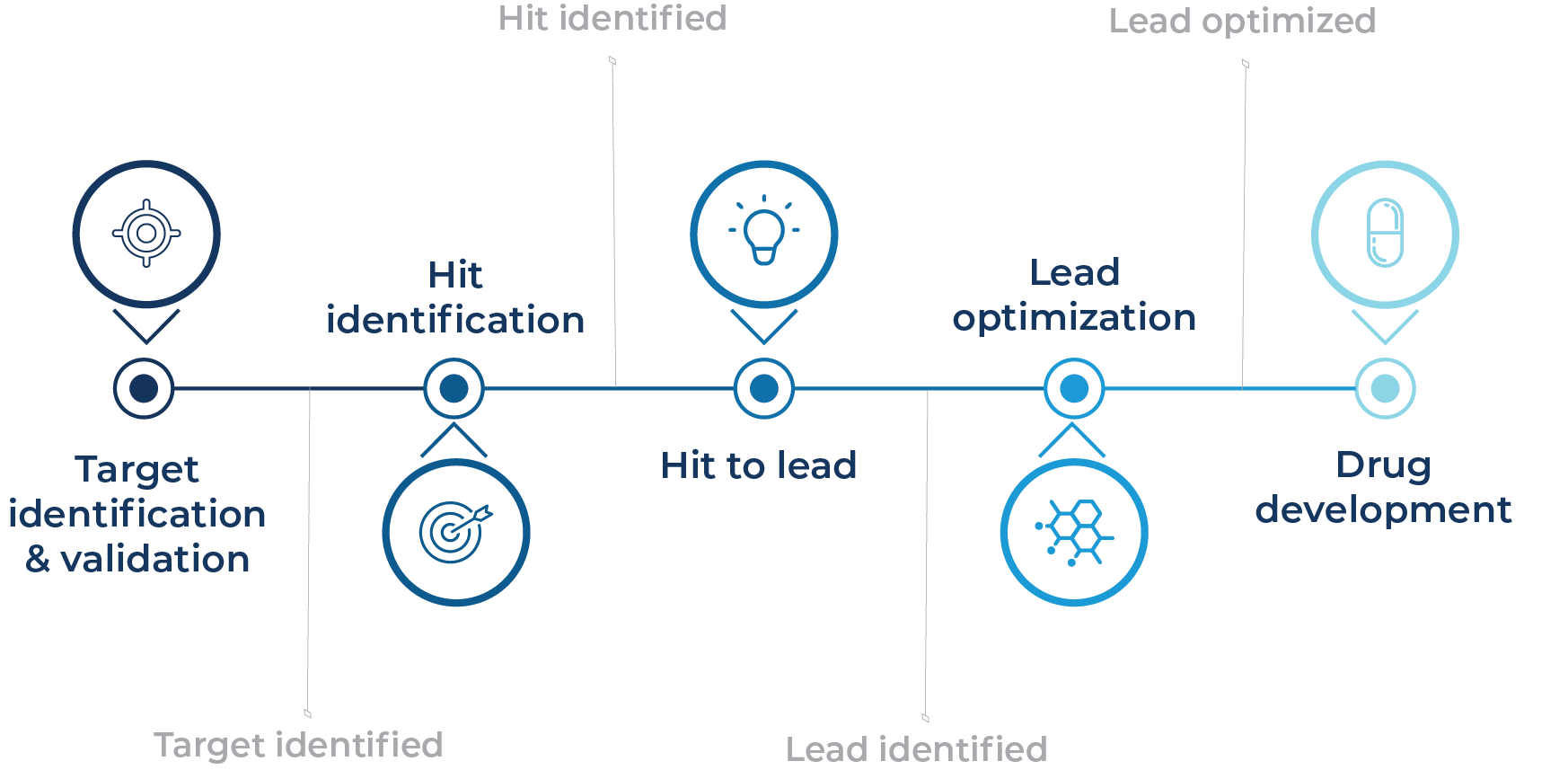
A scalable and cost-effective transcriptomic profiling solution that accelerates target identification and validation, hit identification, hit-to-lead, and lead optimization. By providing deep gene expression insights across thousands of compounds, MERCURIUS™ DRUG-seq reveals mechanisms of action, detects on/off-target effects, and uncovers toxicity markers, empowering faster, data-driven decisions throughout the drug development pipeline.
Identify drug resistance mechanisms at the transcriptomic level by tracking pathway rewiring and molecular adaptation, supporting response stratification and follow-up hypotheses.
DRUG-seq helps surface early toxicity and stress signals at screening scale, making it a powerful fit for NAM-based toxicology workflows and NGRA study designs.
The examples below show how transcriptome-wide readouts can reveal mechanisms of action, detect combination effects, and generate actionable insights across large screening campaigns.
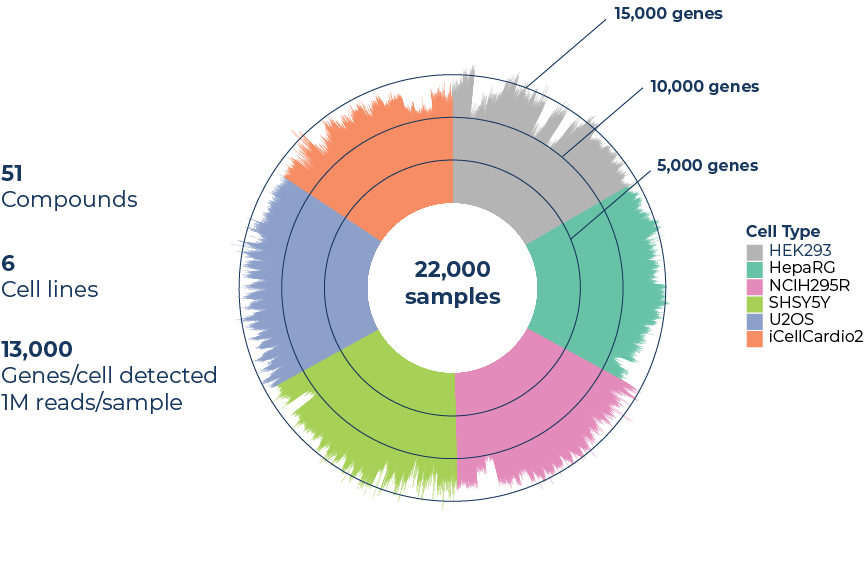
For large scale studies
Gene detection overview of a case study on 22,000 samples and 51 compounds in four doses, on 6 cell lines, sequenced at 1M reads per sample.
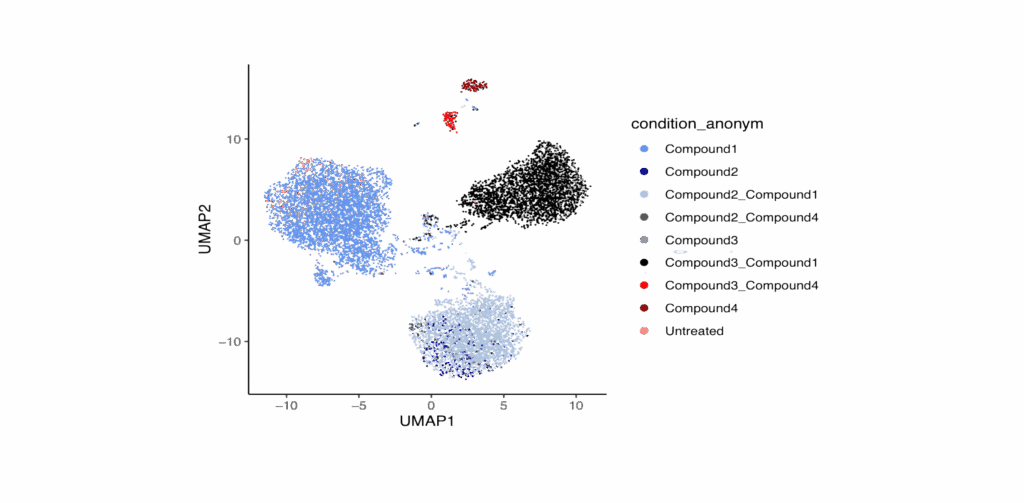
Clustering and co-clustering
Use unbiased gene expression measurements to support accurate compound clustering and co-clustering analyses.
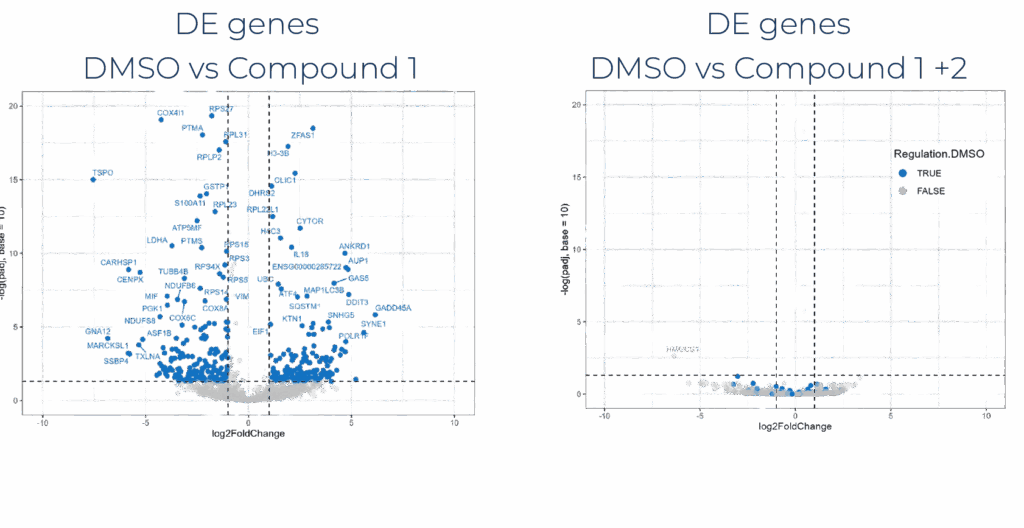
Drug combinations
Detect synergistic or antagonistic combination effects from transcriptomic signatures.
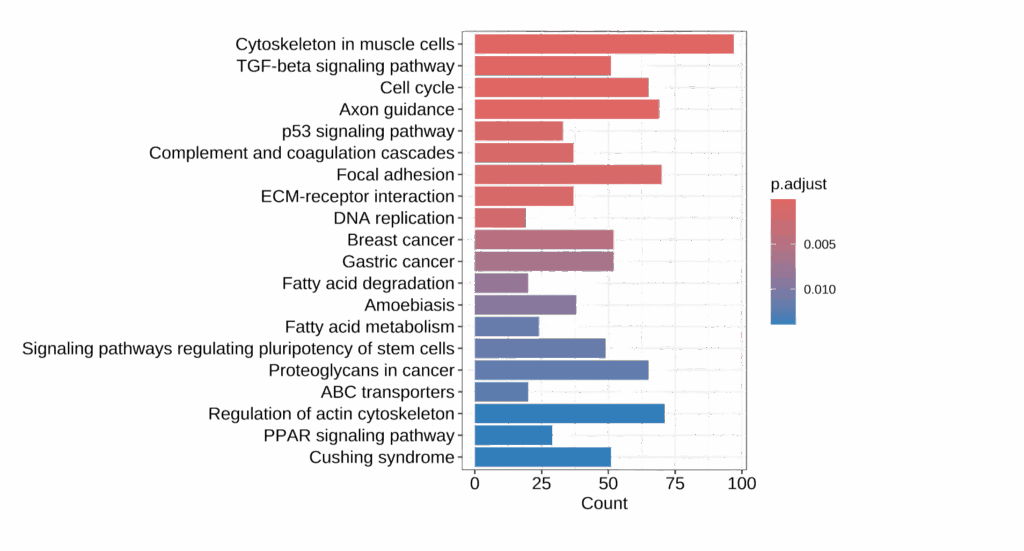
Enables detailed pathway analysis of biological processes
Interrogate the molecular pathways and identify altered biological processes
Start with a focused subset to validate the workflow and choose the right DRUG-seq readout (3′ or Total). The pilot can be executed in our labs as a service, or in your lab using MERCURIUS™ DRUG-seq kits.
Validate the workflow on a focused subset and review library QC, sequencing QC, signal-to-noise, reproducibility, and sequencing depth before scaling.
Expand into the full matrix of models, doses, time points, and combinations, while keeping controls and reporting consistent across plates and batches.
DRUG-seq
Total DRUG-seq
• Full-length total RNA sequencing
• Early multiplexing of up to 384 samples
• Directly from cell lysates without prior RNA isolation
• Ideal for comprehensive genome-wide gene expression profiling of coding and non-coding transcripts for isoform detection, alternative splicing analysis, and fusion gene detection

DRUG-seq Service
• DRUG-seq library prep
• Next Gen Sequencing
• Data pre-processing
Multimodal Service
• Cell Painting
• DRUG-seq library prep
• Next Gen Sequencing
• Data pre-processing

Complete Service
• Cell Culture
• Cell Painting
• DRUG-seq library prep
• Next Gen Sequencing
• Data pre-processing


For the first time, 1536-well whole transcriptome profiling brings biologically rich readouts to primary screening and large-scale perturbation dataset generation. In this webinar, Alithea Genomics…
June 8, 2026 Alithea Genomics, a pioneering biotech company specializing in scalable RNA sequencing solutions, today announced a new agreement with Revvity Inc. to broaden…
Biomarker discovery using gene expression signatures has transformed pharmaceutical discovery and development, from early hit triage and target validation to toxicology, dose optimization, and patient…
Next-generation AI models for drug discovery are only as powerful as the quality, size, and perturbation-richness of their training data. Yet no publicly available dataset…
MERCURIUS™ DRUG-seq is a transformative tool for compound screening and drug discovery, combining unbiased, ultra-high-content, and high-throughput compound screening with massively parallel and extraction-free transcriptomics. This metho d uses highly optimized and rigorously evaluated sample barcodes and unique molecular identifiers to tag the 3’ poly(A) tail of all mRNA molecules in a sample-specific manner during the first-strand synthesis step of cDNA library preparation.
The DRUG-seq protocol is designed to work with frozen cells and bypass RNA extraction. Thanks to the highly optimized lysis buffers for cell lysis of 2D cell cultures and organoid models, it efficiently generates library preps without prior RNA isolation.
Unlike the original DRUG-seq protocol, which relies on the use of template-switching oligos (TSO) and pre-amplification for the second-strand cDNA synthesis, the MERCURIUS™ DRUG-seq streamlines the workflow by performing the reverse transcription with a high-yield enzyme, thus eliminating the potential biases and duplication introduced with pre-amplification.
This optimized workflow not only simplifies the protocol but also delivers superior performance: higher mapping and gene detection rates.
MERCURIUS™ DRUG-seq stands out from traditional RNA-seq methods by offering a high-throughput, cost-efficient, and streamlined workflow specifically designed for large-scale screening applications. One of the key differences lies in its sample multiplexing strategy: DRUG-seq allows multiple RNA samples to be barcoded and pooled at the earliest step of the protocol—right after cell lysis—so that the entire library preparation can proceed in a single tube. This significantly reduces both hands-on time and reagent costs.
In contrast, standard RNA-seq workflows typically require individual RNA extraction and processing for each sample, followed by separate library preparations. This makes them more labor-intensive, costly, and less scalable, especially when working with large numbers of conditions, compounds, or replicates—common in drug discovery pipelines.
MERCURIUS™ DRUG-seq is used for:
Drug discovery: Identifying new drug candidates and understanding how existing drugs work at a molecular level.
Mechanism of action studies: Understanding how drugs affect cellular pathways and gene regulation.
Drug repurposing: Identifying new uses for existing drugs based on gene expression changes.
Personalized medicine: Tailoring treatments based on individual genetic responses to drugs.
MERCURIUS™ DRUG-seq can be performed on various sample types, including: Cell lines, Primary cells, Organoids or Spheroids.
Validated cells lines
| Cell line/Tissue | Organism |
| HT1080 SMARCA4 KO | Human |
| hTEC | Human |
| iPSC microglia | Human |
| MCF7 | Human |
| HepG2_IGF1RKO | Human |
| Beas-2B | Human |
| dTHP-1 | Human |
| iPSC derived cardiomyocytes | Human |
| Endothelial | Human |
| Hepatocyte | Human |
| HEPG2 | Human |
| Hepatocyte | Human |
| SF9 | Spodoptera |
| A549 | Human |
| Breast Cancer (MCF7) | Human |
| B lymphoblast MM.1s. | Human |
| Hek293 | Human |
| Cell line/Tissue | Organism |
| HeLa Cells | Human |
| U2-OS | Human |
| Hek293 | Human |
| dTHP-1 | Human |
| AsPC-1 | Human |
| PBMC (TCD4) | Human |
| Skeletal muscle (LHCN-M2) | Human |
| HepaRG | Human |
| Macrophage (MV-4-11) | Human |
| PBMCs (Blood) | Human |
| Microglia | Human |
| Fibroblast Like Synoviocyte donor 1502 | Human |
| Fibroblast | Human |
| iPSC-derived neuron | Human |
| Human lung carcinoma epithelial cell line | Human |
| Human breast epithelial adenocarcinoma cell line | Human |
| Human liver hepatocellular carcinoma cell line | Human |
| Human cardiomyocytes derived from induced pluripotent stem cells (iPSCs) | Human |
We require the cells/organoids to be washed in PBS to avoid interference with our proprietary lysis and reverse transcription.
The recommended range of input material is in the range of 5’000-50’000 cells.
Tell us about your project and we will help you find the right approach.