Ultra-sensitive, full-length, and plate-based single-cell and low-input RNA-seq technology for sorted cells or low-input RNA samples
MERCURIUS™ FLASH-seq is built for studies where sensitivity and transcript-level detail matter simultaneously.
Based on the method first published in Nature Biotechnology, MERCURIUS™ FLASH-seq combines a plate-based workflow with full-length transcript coverage and is suitable for sorted single cells, rare populations, and precious low-input samples. Instead of choosing between depth and practicality, you can generate rich transcriptomic data in a workflow that is fast, scalable, and compatible with automation.

High sensitivity helps recover low-abundance transcripts and retain signal even in rare or heterogeneous populations, giving you a clearer view of cellular diversity and state.
Full-length coverage supports differential gene expression, alternative splicing, and isoform analysis in the same experiment, so you can move beyond counting genes to understanding transcript structure and function.
A plate-based workflow integrates naturally with FACS and automation, reduces hands-on time, and makes high-resolution single-cell and low-input RNA-seq more practical for routine studies.
Dissect tumor heterogeneity by sequencing rare cancer stem cells or circulating tumor cells. MERCURIUS™ FLASH-seq offers the resolution and sensitivity to enable detection of expressed mutations, splicing isoforms, and gene fusions, providing deep insights into clonal evolution and therapeutic resistance in individual cancer cells.
By mapping gene expression trajectories across individual cells during early development, MERCURIUS™ FLASH-seq allows researchers to study how cells differentiate and form tissues, helping to unravel the complex processes that drive organismal development.
MERCURIUS™ FLASH-seq can map the transcriptomic diversity of brain cell types, helping uncover neuronal functions and mechanisms underlying neurological disorders. It has also been used to sequence rare, sorted retinal and brain cells, as well as sub-cellular content.
MERCURIUS™ FLASH-seq tracks gene expression in immune cells, revealing responses to infections, autoimmune disorders, and other diseases. TCR chains and CDR3 regions can be inferred directly from single-cell FLASH-seq data, which also captures intracellular viral transcriptomes such as retroviruses and AAVs for host–virus studies.
The sensitivity of MERCURIUS™ FLASH-seq makes it efficient even with sub-cellular amounts of RNA (<1 pg), such as exosome preparations or biopsies of individual cells.
MERCURIUS™ FLASH-seq also detects transcriptomes from intracellular viruses such as retroviruses and adeno-associated viruses (AAVs), enabling direct study of host–virus interactions at single-cell resolution.
Researchers can use MERCURIUS™ FLASH-seq to evaluate the effect of antisense oligonucleotides (ASOs) on target splicing at single-cell resolution. This enables precise tracking of treatment efficacy across rare or responsive cell subsets within a population.
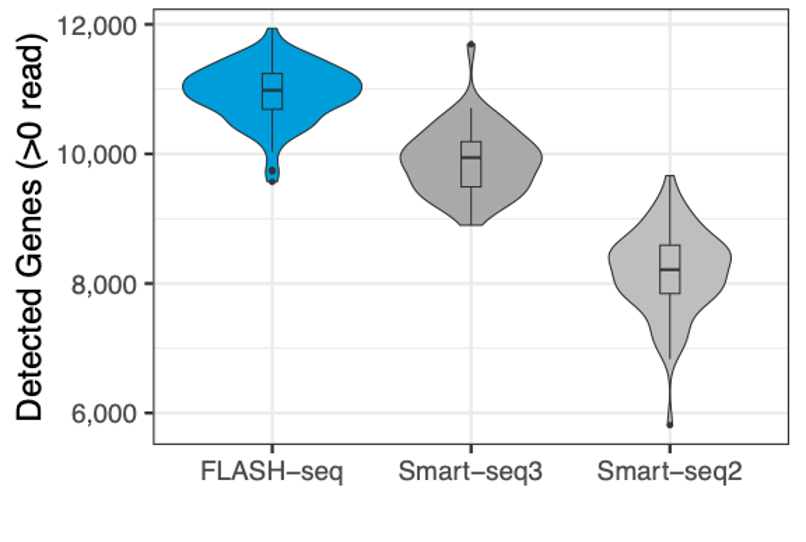
MERCURIUS™ FLASH-seq shows the highest sensitivity in gene detection
The number of detected genes in HEK 293T cells processed with different protocols. Reads were downsampled to 500,000 raw reads.
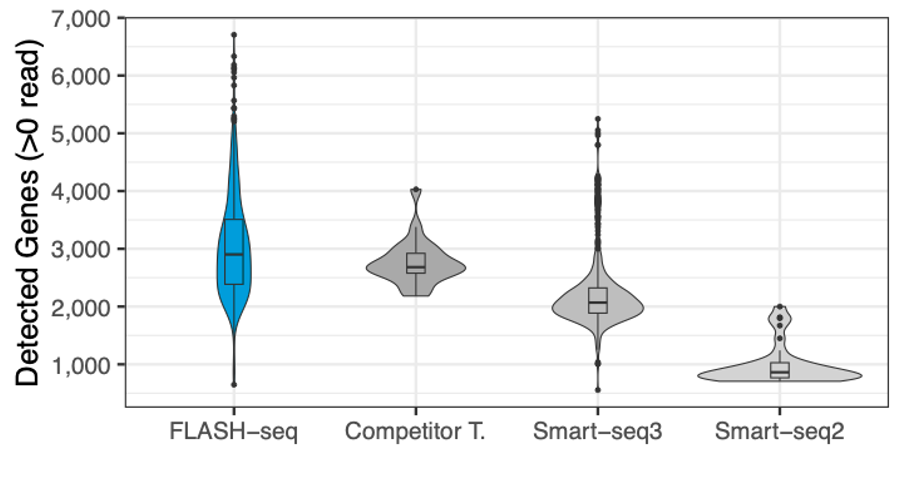
The sensitivity of MERCURIUS™ FLASH-seq is retained even in highly heterogeneous populations
Number of genes detected in human PBMCs, processed with different protocols, and the number of reads downsampled to 125,000 raw reads.
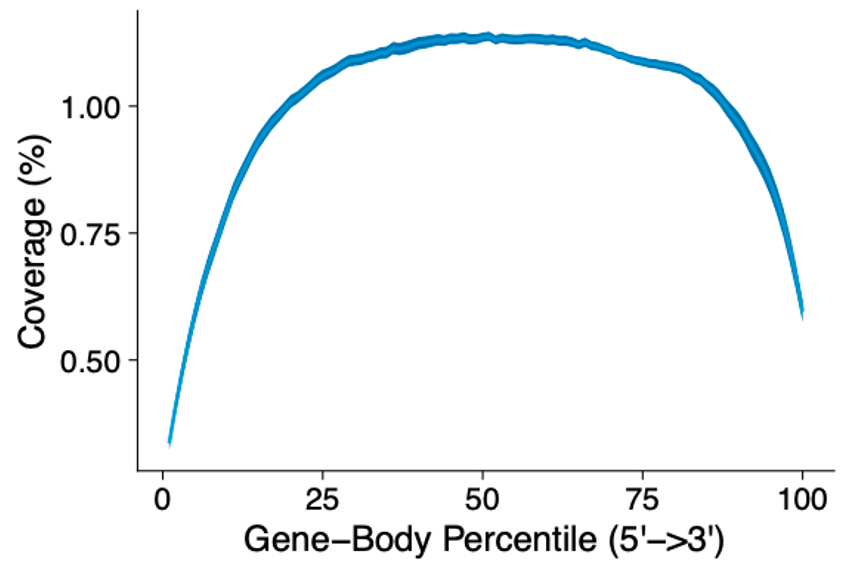
MERCURIUS™ FLASH-seq is a full-length scRNA-seq protocol
Gene body coverage shows a uniform read distribution across the entire gene body for the FLASH-seq protocol.
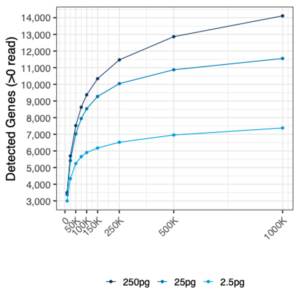
MERCURIUS™ FLASH-seq shows high sensitivity for low sample inputs
The number of genes detected in HEK 293T cells using different RNA inputs (from 2.5 pg to 250 pg) at different sequencing depths.
Single-cell
FLASH-seq kit
• Single-cell, full-length mRNA sequencing
• Ultra-sensitive and rapid
• Plate-based
• Directly from cell lysates without prior RNA isolation
• Ideal for rare cell capture and isoform detection
Low-input
FLASH-seq kit
• Early multiplexing of up to 96 samples
• Up to 2x more genes detected than other commercially available solutions. Ideal for rare cell capture.
• Low RNA inputs from precious samples (1 pg to 1 ng per well).

Single-cell
FLASH-seq service

Low-input
FLASH-seq service


Single-cell RNA sequencing (scRNA-seq) has revolutionized our ability to examine the cellular heterogeneity of complex samples, identify new cell types, and explore transcriptional regulation at…
The Smart-seq2 protocol was the gold standard for full-length plate-based single-cell RNA-seq, offering superior sensitivity and the transcript coverage necessary to detect splice isoforms, allelic…
Lausanne, Switzerland and Frederick, MD – June 19, 2025 – Alithea Genomics, a pioneering biotech company based in Switzerland and the USA specializing in high-throughput…
MERCURIUS™ FLASH-seq is a plate-based method that focuses on polyadenylated RNA, which includes most mRNAs. It provides detailed information on gene structure and alternative splicing. Its ability to capture full-length mRNA transcripts and detect low-abundance genes makes it a powerful tool for gene expression profiling.
FLASH-seq protocol was designed to enhance both the sensitivity and efficiency of single-cell mRNA sequencing compared to the most famous plate-based method, Smart-seq2
Read more about the technology in our blog posts: here and here.
We have significantly enhanced the original FLASH-seq method to offer a streamlined workflow and superior data output. This plate-based technology (available in 96- and 384-well formats) features a novel, non-toxic tagmentation buffer and delivers ultra-sensitive gene detection, capturing up to two times more genes compared to other commercially available solutions.
The MERCURIUS™ FLASH-seq protocol is fully compatible with whole FACS-sorted cells.
We do not recommend very large cells, such as the cardiomyocytes, as they are not compatible with the FACS sorting step.
We require the cells to be directly FACS-sorted in the dedicated well plates. The 96- and 384-plates contain the lysis buffer. It is important for the user to sort the cells in the middle of the well. Please refer to the User Guide and the Sample Submission Guidelines for more details.
The average recommended sequencing depth is 250’000 reads/cell.
Tell us about your project and we will help you find the right approach.