
Single-cell RNA sequencing (scRNA-seq) has created a new era of understanding cellular diversity by allowing gene expression analysis in individual cells, revealing details that are masked when studying bulk cell populations. This fine-tuned resolution is critical for unraveling cellular functions, states, and interactions, especially in complex tissues or disease environments.
Among the most prominent methods for scRNA-seq are FLASH-seq (1) and Vast Transcriptome Analysis of Single cells by dA-tailing (VASA-seq) (2), which offer unique approaches and capabilities. While both methods push the boundaries of transcriptomics, each has its own set of limitations and challenges. Let’s explore how these technologies compare and their potential applications.
Overview of FLASH-seq
FLASH-seq is a plate-based method that focuses on polyadenylated RNA, which includes most mRNAs. It provides detailed information on gene structure and alternative splicing. Its ability to capture full-length mRNA transcripts and detect low-abundance genes makes it a powerful tool for gene expression profiling. FLASH-seq protocol was designed to enhance both the sensitivity and efficiency of single-cell mRNA sequencing compared to the most famous plate-based method, Smart-seq2(3).
Workflow at a glance: In the first step of the workflow, individual cells are sorted into plates containing lysis buffer. Upon contact with the lysis buffer, cells are broken open, releasing their RNA, which is then captured using oligo(dT) primers that bind to the poly(A) tails of mRNA molecules. The captured RNA is reverse-transcribed into complementary DNA (cDNA) and amplified within a single RT-PCR reaction. cDNA molecules are fragmented and PCR amplified using cell-specific indexes. FLASH-seq integrates a “low-amplification” version of the protocol where cDNA molecules can be directly fragmented without any prior purification or dilution. High-throughput sequencing platforms are used to analyze the prepared libraries, yielding data that is then processed to quantify gene expression and uncover alternative splicing events (Figure 1).
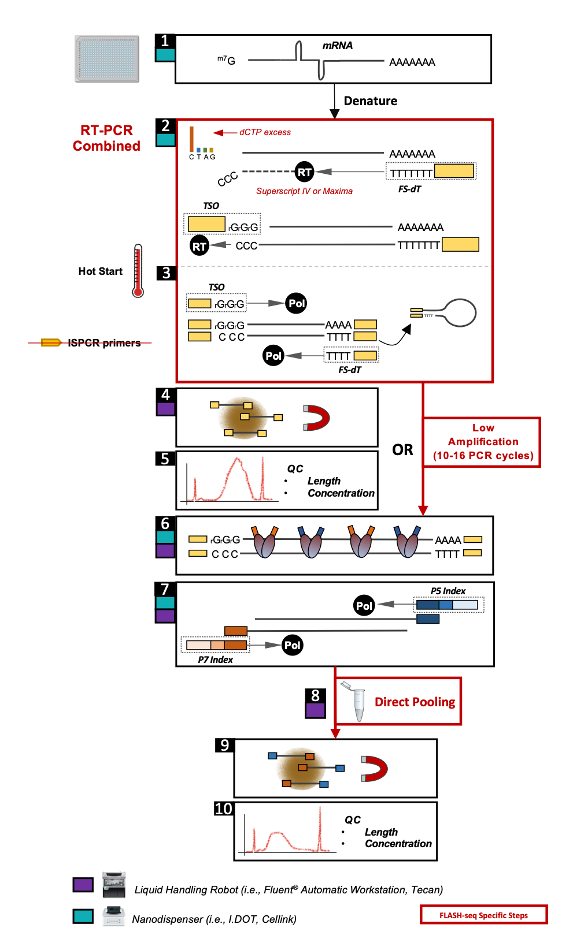
Figure 1- Overview of the FLASH-seq single-cell molecular workflow..Taken from (1).
Overview of VASA-seq
VASA-seq, on the other hand, captures both non-polyadenylated and polyadenylated transcripts across their length in plate and droplet microfluidic formats. This comprehensive view allows researchers to study a broader range of RNA species, including regulatory non-coding RNAs, which play important roles in gene expression control. In essence, VASA-seq was developed to combine excellent sensitivity, full-length coverage of total RNA, and high throughput.
Workflow at a Glance: The VASA-seq protocol begins with the fragmentation of RNA molecules from the single-cell lysate, followed by end repair and poly(A) tailing to facilitate cDNA synthesis using barcoded oligo-dT probes. A unique fragment identifier (UFI) is incorporated to ensure accurate molecule quantification with strand specificity. Barcoded cDNA is then amplified through in vitro transcription, and ribosomal RNA (rRNA) is depleted. In the final step, the libraries are amplified using unique dual-indexed (UDI) PCR primers, which help detect index hopping during Illumina sequencing, followed by sequencing (Figure 2).
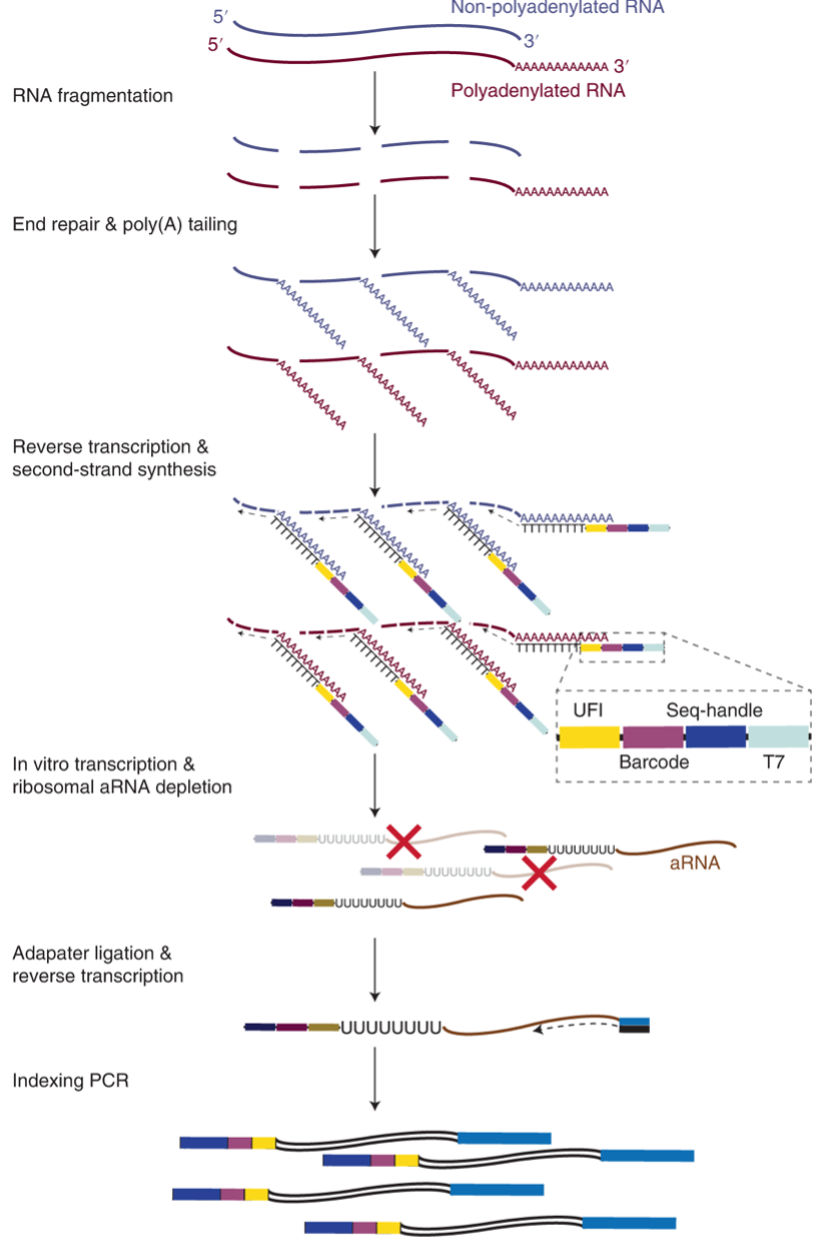
Figure 2- Overview of the VASA-seq single-cell molecular workflow. Taken from (2).
Comparative Analysis: FLASH-seq vs. VASA-seq
Understanding the strengths and limitations of each method allows researchers to make informed decisions when selecting the most suitable method for their studies. By carefully balancing their research needs with the challenges presented by each technique, scientists can maximize the insights gained from their single-cell RNA sequencing experiments, pushing the boundaries of our knowledge of gene expression and cellular function.
|
General considerations |
FLAS-seq |
VASA-seq |
|
RNA coverage |
Captures polyadenylated RNA, focusing on mRNAs and splicing |
Sequences both polyadenylated and non-polyadenylated RNA, providing a more inclusive view of the transcriptome. |
|
Sensitivity and specificity |
Highly sensitive in detecting polyadenylated RNAs and alternative splicing (4). |
Equally sensitive, but with the added benefit of capturing non-coding RNAs, offering insights into gene regulation (4). |
|
Applications |
Ideal for studies that focus on gene expression profiling, alternative splicing, and mRNA diversity. |
Suitable for comprehensive transcriptome analysis, particularly in studies involving non-coding RNAs and gene regulation. |
Challenges and Limitations
FLASH-seq:
- Polyadenylated RNA Focus: Since it targets only polyadenylated RNA, FLASH-seq misses out on important non-coding RNAs that are critical for understanding gene regulation.
- Cost and Scalability: FLASH-seq can be expensive, especially when scaled up, which may limit its use in large studies.
- Technical Complexity: Handling FLASH-seq protocols requires precision and optimization, which can increase the technical burden on labs.
VASA-seq:
- Data Complexity: The broader range of RNA sequenced by VASA-seq increases data complexity, requiring advanced bioinformatics tools and expertise for analysis.
- RNA Degradation Sensitivity: VASA-seq is more sensitive to RNA degradation, which can compromise data quality, making high-quality RNA samples essential.
- Technical Variability: Results from VASA-seq can vary depending on the protocol, making reproducibility a challenge across different labs.
Common Challenges:
- Single-Cell Isolation: Both methods rely on efficient cell isolation, which can be technically demanding and may lead to cell loss, biasing the results.
- Data Analysis: Handling the large datasets generated by both methods requires sophisticated bioinformatics pipelines, which can be resource-intensive.
- Batch Effects and Technical Noise: Both techniques can suffer from variability introduced by batch effects, making careful experimental design and data normalization crucial.
Applications
The potential of both FLAS-seq and VASA-seq spans across numerous fields of biomedical research.
- FLASH-seq: offers remarkable potential across various areas of biomedical research, especially in studying rare cell populations that are otherwise difficult to analyze. In cancer research, FLASH-seq’s sensitivity and resolution can reveal cellular heterogeneity, identifying resistant cells contributing to relapse and uncovering new therapeutic targets. In developmental biology, it could be used to map the gene expression patterns across individual cells during early development, providing insights into cellular differentiation (5). Neuroscience benefits from FLASH-seq by enabling the mapping of the brain’s diverse cell types, aiding in the understanding of neuronal function and the molecular basis of neurological disorders, with successful applications in rare retinal and brain cells, as well as sub-cellular content from PATCH-seq experiments (6). In immunology, FLASH-seq tracks gene expression in immune cells, offering a clearer view of immune responses to infections, autoimmune diseases, and more. Additionally, it can infer TCR chains and CDR3 regions at the single-cell level without extra processing (1). The method’s high sensitivity also makes it effective for analyzing sub-cellular RNA content, such as exosome preparations or single-cell biopsies.
- VASA-seq: is widely used in developmental biology, where it captures both coding and non-coding RNAs during processes like embryogenesis (2). Its ability to capture the entire transcriptome has provided key insights into cell differentiation and tissue development (2). VASA-seq was also used in RNA-protein interaction studies. Using a fusion of the RNA-binding protein FMR1 and the RNA-editing enzyme ADAR, scientists found that the fusion protein assembles into stress granules under oxidative stress, where it modifies RNAs. The mutations introduced by ADAR were then detected using VASA-seq, allowing researchers to map and analyze RNA interactions and localization within stress granules (7). In another application focused on the identification of stress granule RNAs in single cells and Drosophila neurons, VASA-seq contributed to showing that the RNA content of stress granules can be reliably identified not only in bulk cells but also in single Drosophila S2 cells, and in tissues (8).
Conclusion: Weighing the Pros and Cons of FLASH-seq and VASA-seq
Both methods have advantages and disadvantages, which need to be evaluated based on the biological background and proposed application. FLASH-seq is an excellent choice for studies focusing on mRNA diversity and alternative splicing, but its focus on polyadenylated RNA limits its ability to capture non-coding RNAs. On the other hand, VASA-seq provides a comprehensive transcriptome view, including non-polyadenylated RNAs, though it faces challenges regarding data complexity and sensitivity to RNA degradation.
References:
- Hahaut, V., Pavlinic, D., Carbone, W. et al. Fast and highly sensitive full-length single-cell RNA sequencing using FLASH-seq. Nat Biotechnol 40, 1447–1451 (2022). https://doi.org/10.1038/s41587-022-01312-3
- Salmen, F., De Jonghe, J., Kaminski, T.S. et al. High-throughput total RNA sequencing in single cells using VASA-seq. Nat Biotechnol 40, 1780–1793 (2022). https://doi.org/10.1038/s41587-022-01361-8
- Picelli, S., Faridani, O., Björklund, Å. et al. Full-length RNA-seq from single cells using Smart-seq2.Nat Protoc 9, 171–181 (2014). https://doi.org/10.1038/nprot.2014.006
- Hornung BVH, Azmani Z, den Dekker AT, Oole E, Ozgur Z, Brouwer RWW, van den Hout MCGN, van IJcken WFJ. Comparison of Single Cell Transcriptome Sequencing Methods: Of Mice and Men. Genes (Basel). 2023 Dec 16;14(12):2226. doi: 10.3390/genes14122226. PMID: 38137048; PMCID: PMC10743076.
- Chabosseau P, Yong F, Delgadillo-Silva LF, Lee EY, Melhem R, Li S, Gandhi N, Wastin J, Noriega LL, Leclerc I, Ali Y, Hughes JW, Sladek R, Martinez-Sanchez A, Rutter GA. Molecular phenotyping of single pancreatic islet leader beta cells by “Flash-Seq”. Life Sci. 2023 Mar 1;316:121436. doi: 10.1016/j.lfs.2023.121436. Epub 2023 Jan 25. PMID: 36706832.
- Marx, V. Patch-seq takes neuroscience to a multimodal place. Nat Methods 19, 1340–1344 (2022). https://doi.org/10.1038/s41592-022-01662-5
- Van Leeuwen W., VanInsberghe M., Battich N., Salmén F., van Oudenaarden A., Rabouille C. Identification of the stress granule transcriptome via RNA-editing in single cells and in vivo. Cell Rep Methods. 2022 Jun 20;2(6):100235. https://pubmed.ncbi.nlm.nih.gov/35784648/
- van Leeuwen W, VanInsberghe M, Battich N, Salmén F, van Oudenaarden A, Rabouille C. Identification of the stress granule transcriptome via RNA-editing in single cells and in vivo. Cell Rep Methods. 2022 Jun 20;2(6):100235. doi: 10.1016/j.crmeth.2022.100235. PMID: 35784648; PMCID: PMC9243631.