

BRB-seq service
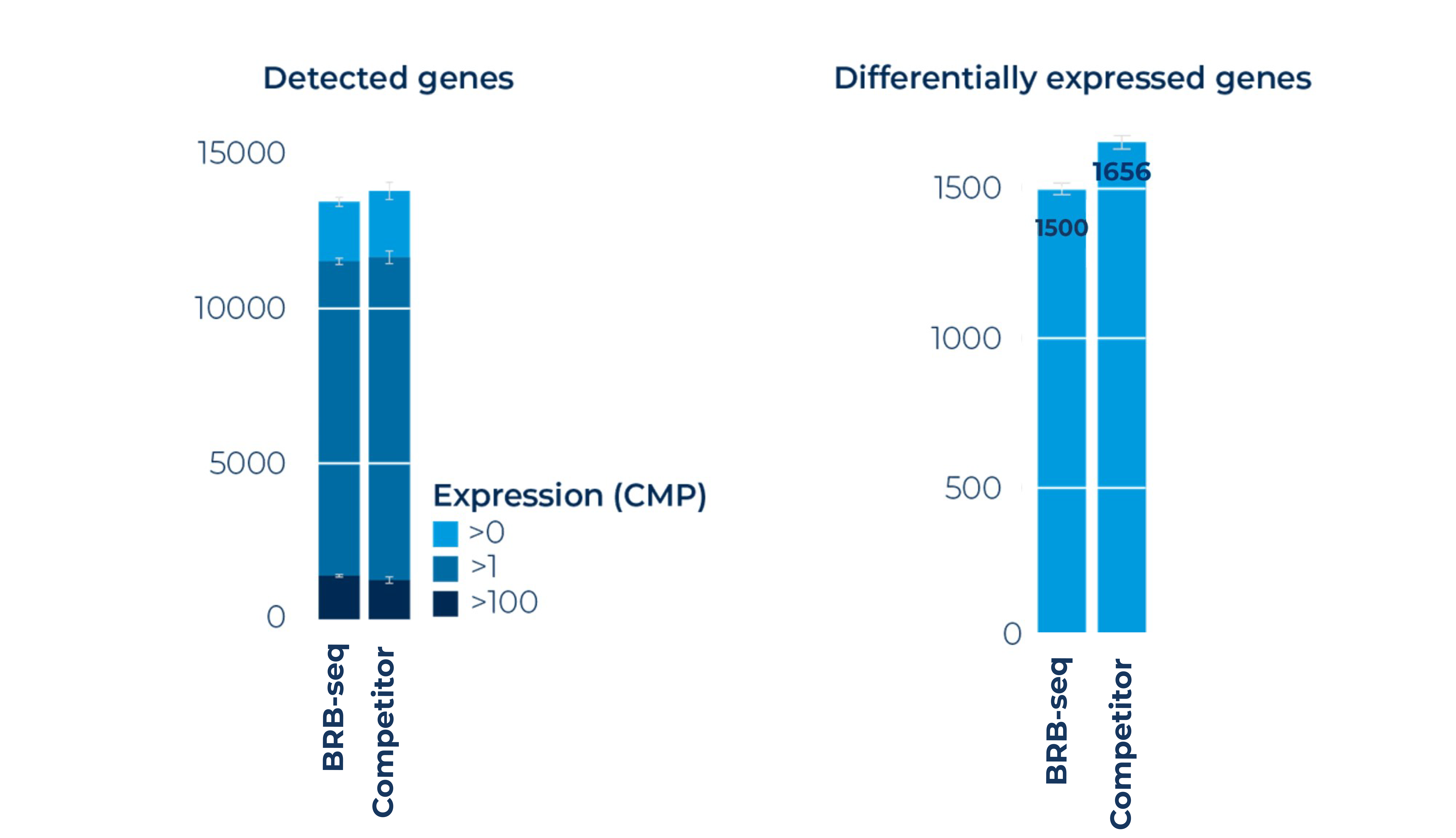
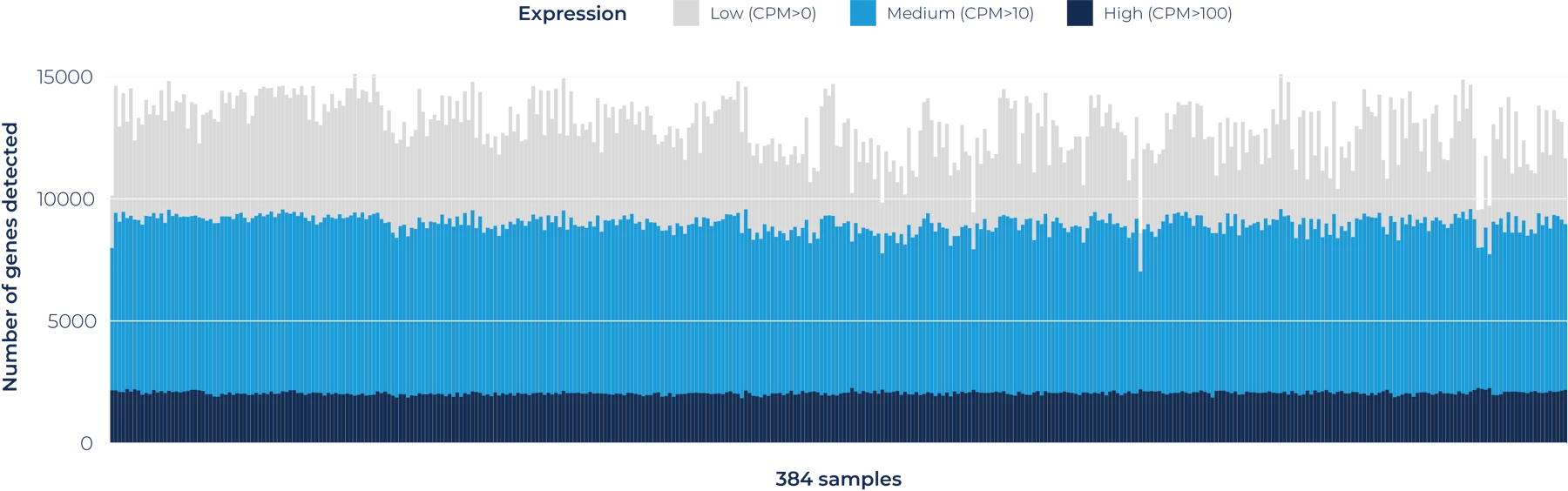
By leveraging BRB-seq, we not only provide industrial and academic clients with quality RNA-seq data, but we also do so with the highest affordability and quick turnaround times.

High-Sensitivity BRB-seq service
This service offers a convenient and streamlined solution for transcriptomics projects with a limited amount of input RNA (starting from 1 ng per sample).

Full-Length BRB-seq service
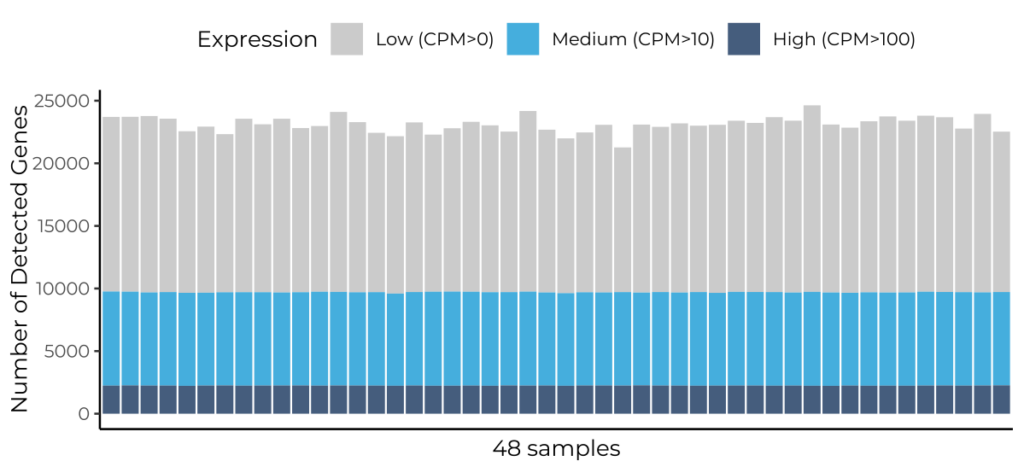
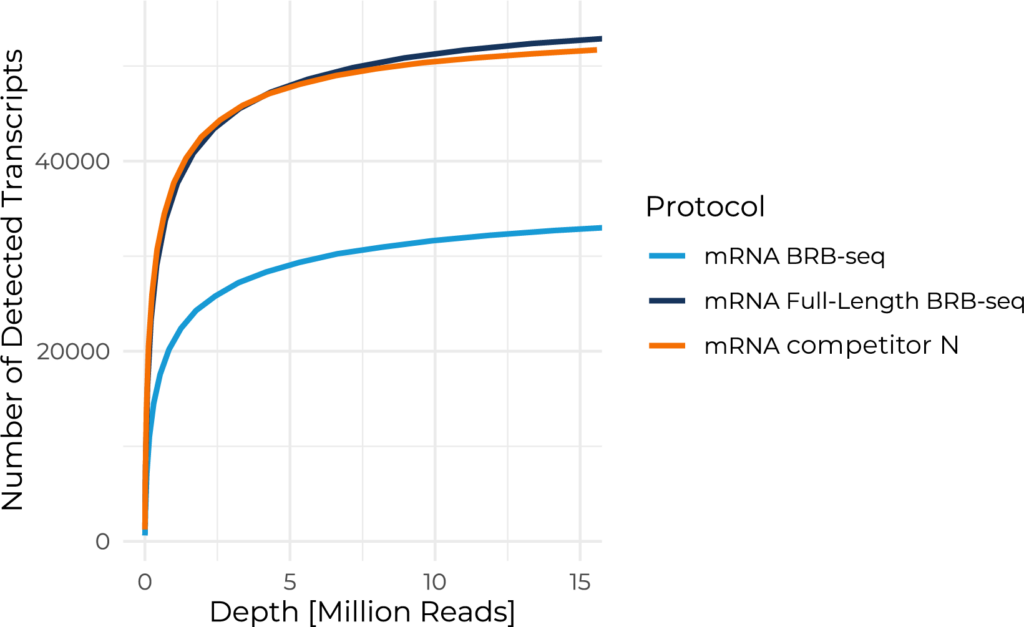
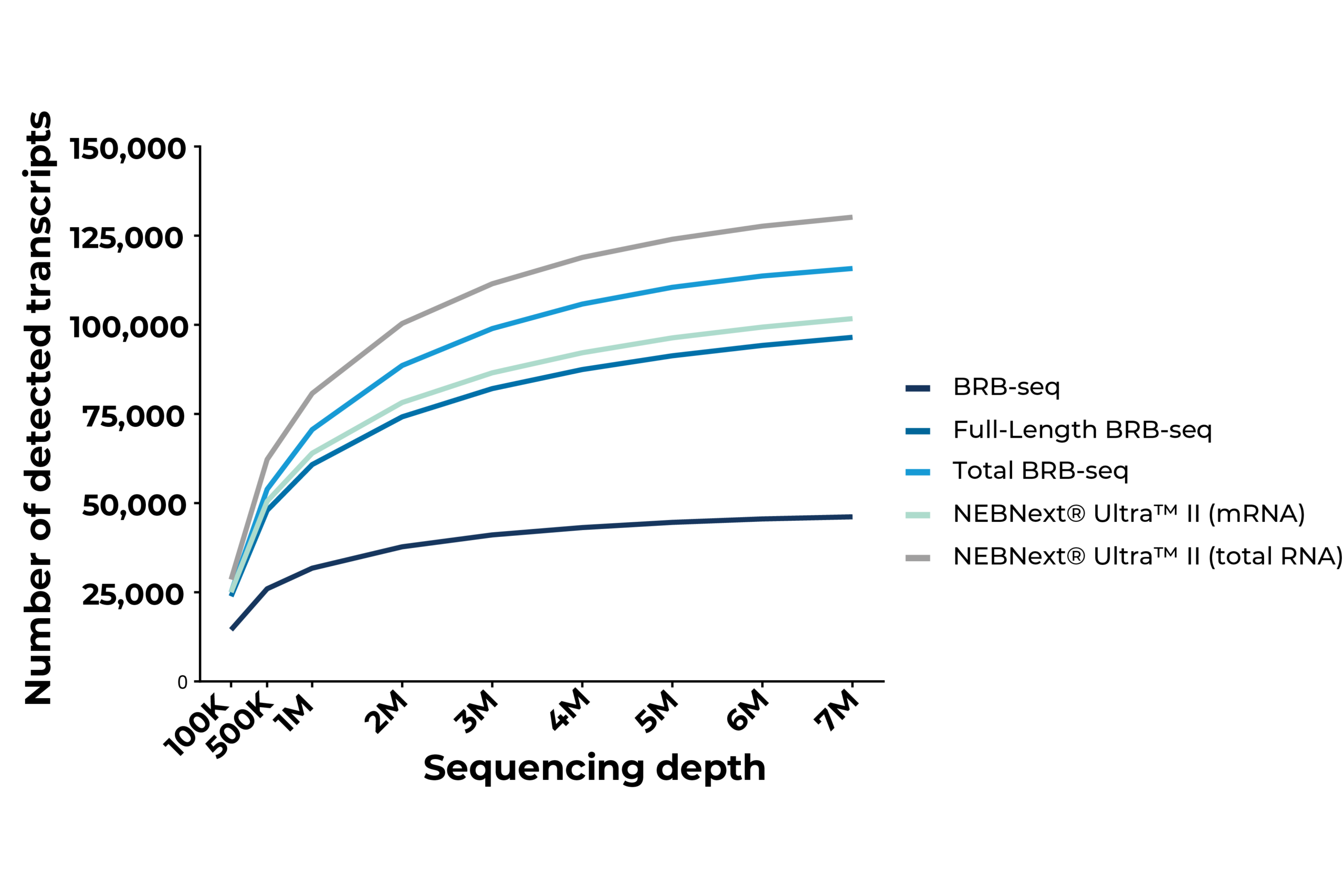
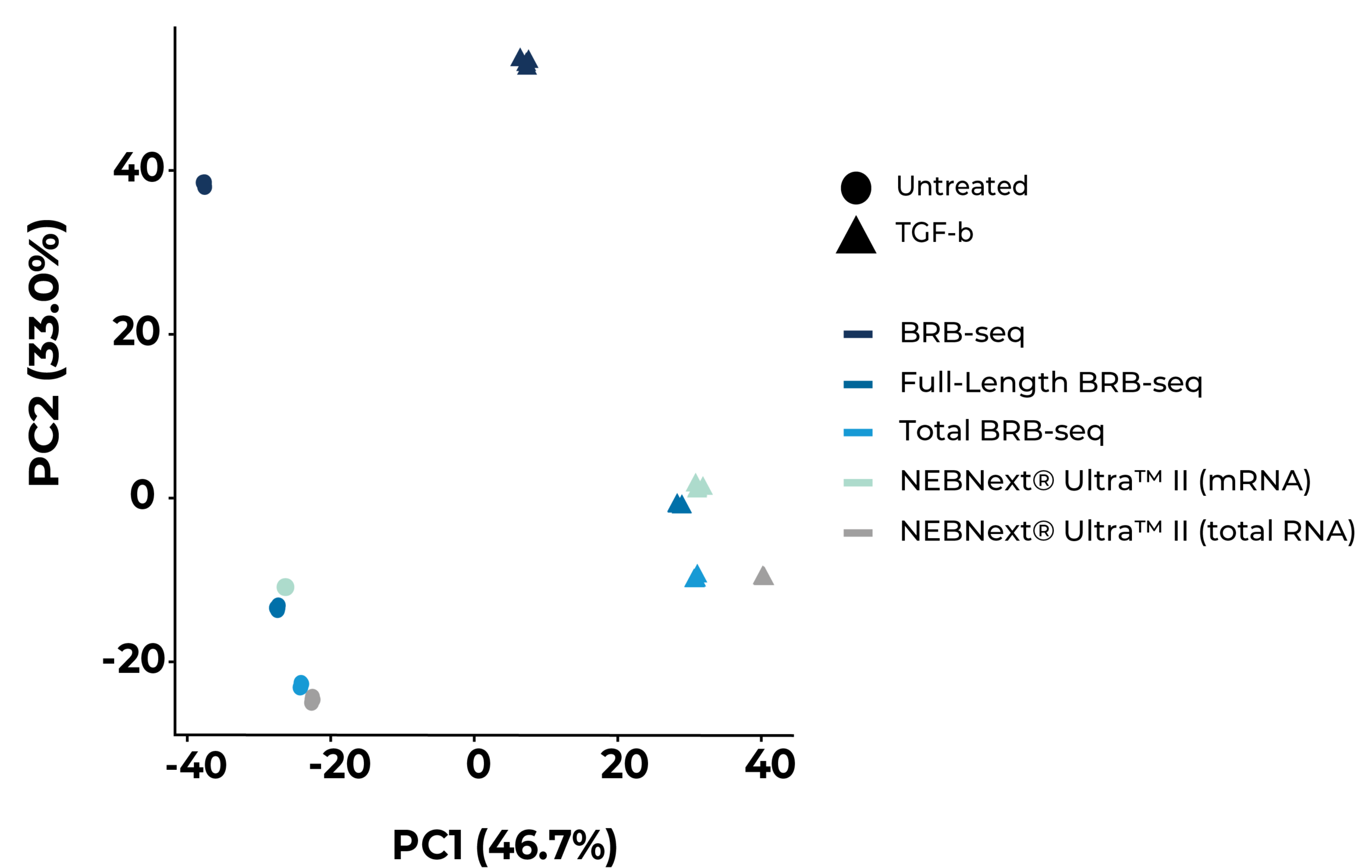
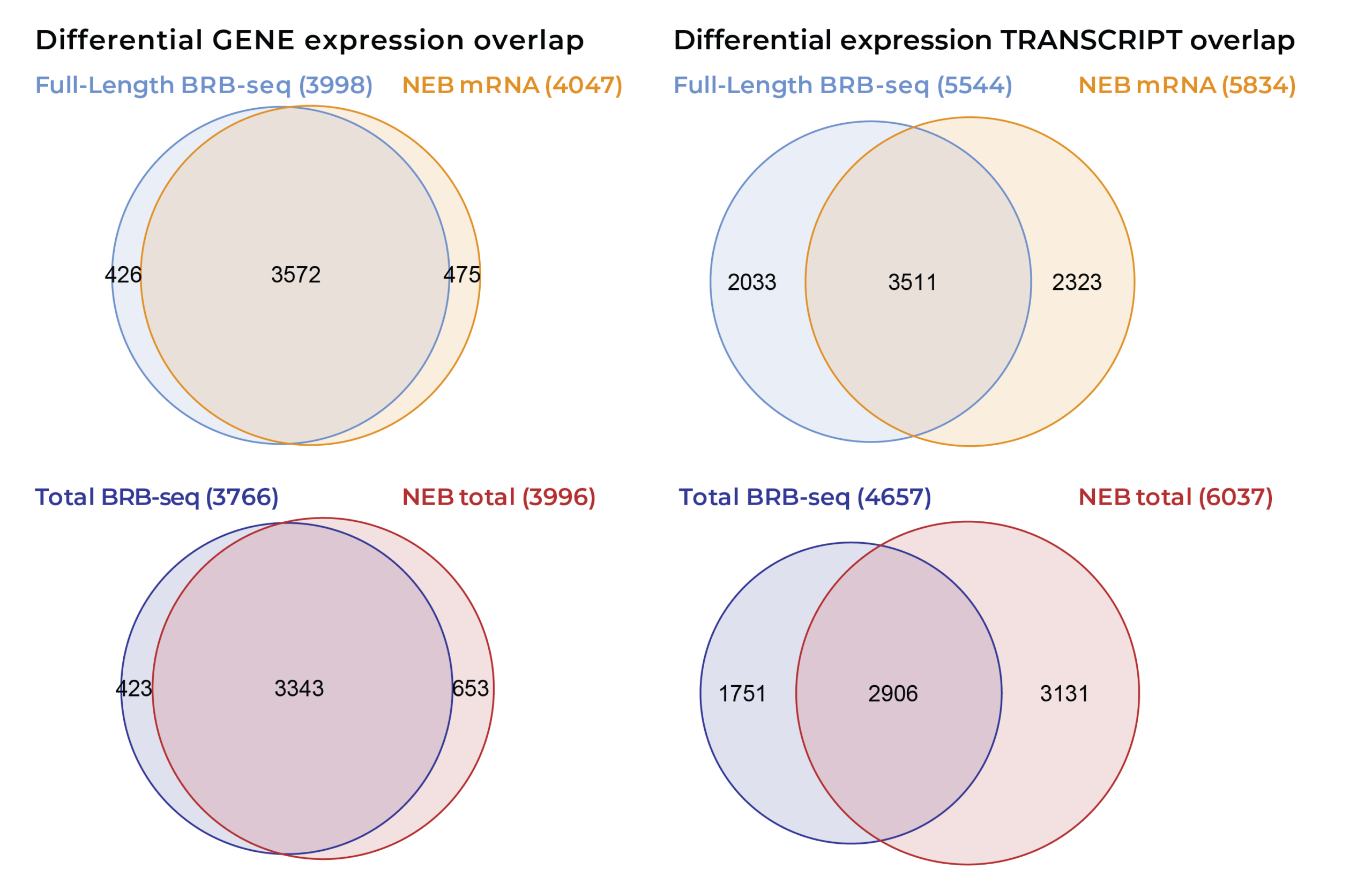
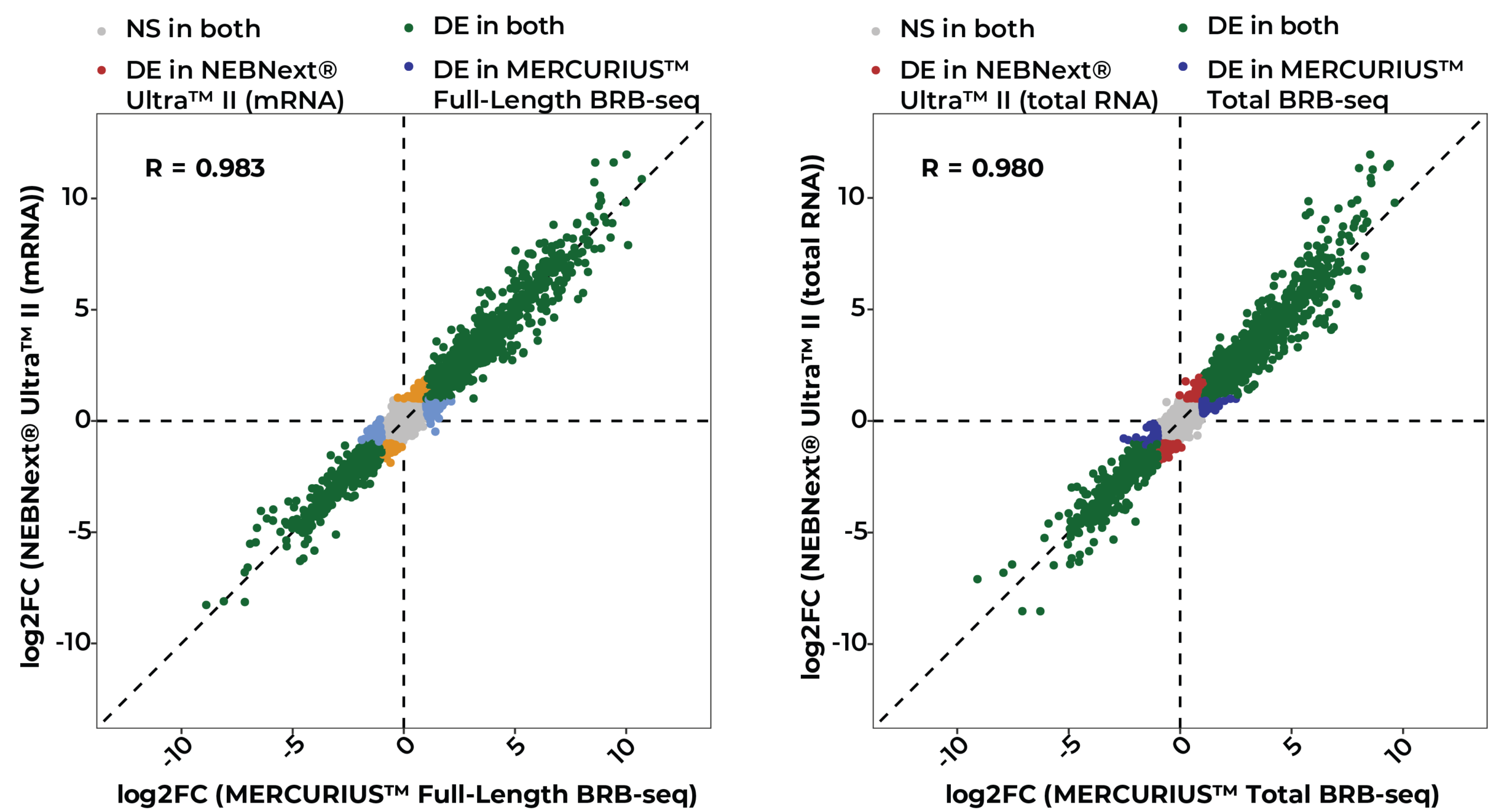
Scalable full-length mRNA sequencing for purified RNA, combining early multiplexing with isoform-level, splicing, and transcript-structure insights.

Total BRB-seq service
The MERCURIUS™ Total BRB-seq service offers a convenient and streamlined solution for full-length total RNA transcriptomics projects of any size.

Blood BRB-seq service
High-throughput 3′ mRNA-seq service for human whole-blood samples, with integrated globin depletion and expertise in high-throughput RNA extraction from PaxGene tubes.

Total Blood BRB-seq service
Multiplexed total RNA-seq service for human whole-blood samples, with inline globin depletion and high-throughput RNA extraction from PaxGene tubes.

