Discover Sample Datasets
HepG2, Hap1, HeLa

MERCURIUS™ DRUG-seq is an extraction-free DRUG-seq kit for drug discovery teams that need transcriptome-wide gene expression readouts at a screening scale.
Designed for ultra-high-throughput detection of protein-coding transcripts in 2D cell lines and primary cells without expensive, time-consuming RNA extraction. Ideal for screening studies with larger cohorts, more conditions, or more timepoints than possible to realistically assess with standard bulk RNA-seq methods.
By combining in-well cell lysis, early barcoding, and multiplexing of up to 96, 384, or 1536* samples in a single tube, MERCURIUS™ DRUG-seq makes high-throughput RNA-seq practical for large compound libraries, multi-dose studies, time-course experiments, and whole-transcriptome CRISPR perturbation screens. The workflow supports compound screening, hit triage, mechanism-of-action studies, toxicity profiling, dose-response experiments, and AI-ready perturbation dataset generation.
* for the 1536 DRUG-seq kit format, go to the dedicated page.
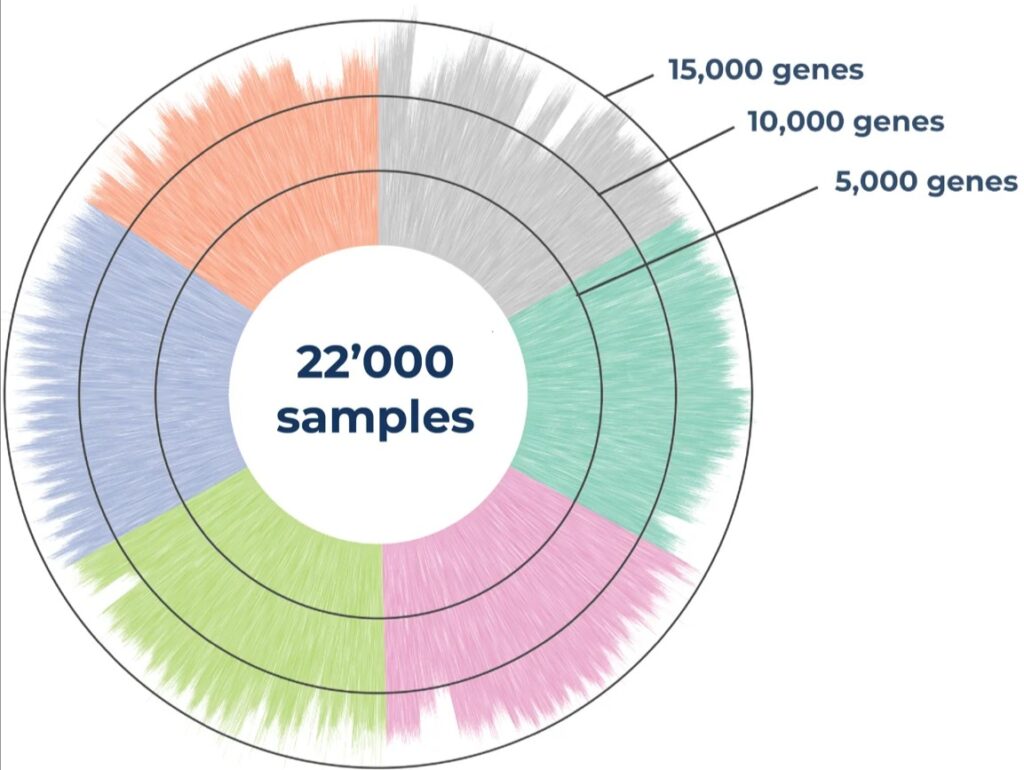
Distribution of the number of detected genes across 22,000 samples in 125x 384-well MERCURIUS™ DRUG-seq plates from six different frozen cell lines. The library was sequenced at an average of 1 million reads per sample on an Illumina NovaSeq 6000.
Compounds
Cell lines
Genes/sample
1M reads/sample
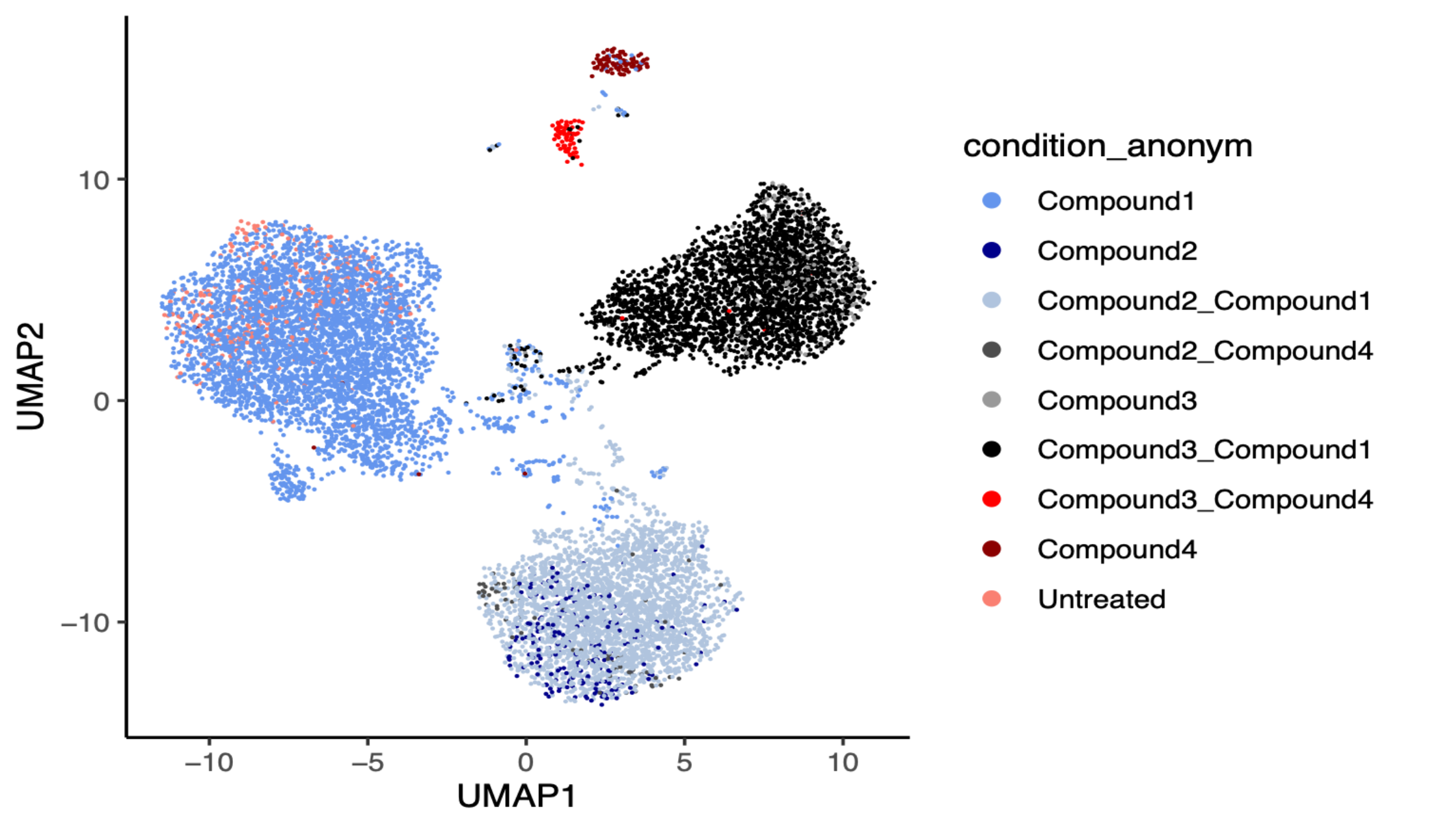
UMAP projection of transcriptional profiles from cells treated with four compounds individually and in combination, alongside untreated controls. Each point represents a single sample, coloured by treatment condition. Samples cluster by transcriptional similarity, revealing that single-compound treatments and combination treatments occupy distinct regions of transcriptional space.
To have access to the deep-sequenced dataset (2 M reads per sample) contact us.
To have access to the deep-sequenced dataset (7.3 M reads per sample) contact us.
To have access to the deep-sequenced dataset (7.1 M reads per sample) contact us.
| HepaRG | Liver |
| HepG2 | Liver (HCC) |
| Huh7 | Liver (HCC) |
| Hep3B | Liver (HCC) |
| PHH (Primary Human Hepatocytes) | Primary liver |
| NCI-H295R | Adrenal/liver metabolism |
| MCF7 | Breast cancer |
| A549 | Lung carcinoma |
| H358 | Lung cancer |
| NCI-H1563 / H1048 | Lung cancer |
| DLD-1 | Colorectal cancer |
| SW837 | Colorectal cancer |
| HCT116 | Colorectal cancer |
| LS180 | Colorectal cancer |
| COLO201 | Colorectal cancer |
| C2BBe / C2BBe1 | Colorectal |
| GP5d | Colorectal |
| U2OS | Osteosarcoma |
| A172 | Glioblastoma |
| PSN-1 | Pancreatic cancer |
| AsPC-1 | Pancreatic cancer |
| SU.86.86 | Pancreatic cancer |
| A375 | Melanoma |
| HaCaT | Keratinocyte |
| UMUC3 | Bladder cancer |
| 5637 | Bladder cancer |
| HT1197 | Bladder cancer |
| Cal29 | Bladder cancer |
| UBLC1 | Bladder cancer |
| PBMC | Primary blood |
| Jurkat | T-cell leukemia |
| Raji | B-cell lymphoma |
| THP-1 | Monocyte |
| U937 | Monocyte |
| MV-4-11 | AML |
| MOLM13 | AML |
| HL60 | Leukemia |
| MM1S | Myeloma |
| KMS12BM | Myeloma |
| CD4+ / CD8+ T cells | Primary immune |
| Tregs / TILs | Immune subsets |
| CD3 T cells | Immune |
| iPSC | Pluripotent |
| iPSC-derived neurons | Neural |
| iPSC-derived cardiomyocytes | Cardiac |
| iPSC-derived cortical neurons | Neural |
| iPSC-derived organoids | Various |
| HMC3 | Microglia |
| Luhmes | Dopaminergic neuron |
| IMR90 | Fibroblast (used in brain spheres) |
| iCell GlutaNeurons | Neurons |
| Astrocytes (human/mouse/rat) | Glial |
| NHDF | Dermal fibroblast |
| MEF | Mouse embryonic fibroblasts |
| HEK293 / HEK293T | Kidney (transformed) |
| ARPE-19 | Retinal epithelium |
| NHEK | Keratinocytes |
| Lung epithelial | Epithelial |
| RPTEC/TERT1 | Kidney (proximal tubule) |
| T84 | Colon epithelium |
| Human adipocytes | Primary |
| Brown adipocytes | Metabolic |
| Visceral adipocytes | Primary |
| Mouse/canine adipose | Animal |
Each DRUG-seq or BRB-seq kit contains reagents (including four pairs of Unique Dual Indexing adapters) sufficient for the complete library preparation process for four different pools.
To note, the total number of RNA samples that can be processed with one kit does not exceed the kit specifications; for instance, a 96-sample kit can be used to prepare up to 96 samples distributed across up to four different libraries.
The DRUG-seq technology can be used to generate high-quality sequencing data from 2000 – 50000 mammalian cells per well. Notably, the kit can be used to pool any number of samples up to the capacity of the provided plate (96 or 384) with two considerations:
• The total cell number per pool should be at least 80000.
• Pooling less than eight samples may result in low-complexity reads during sequencing, decreasing the overall sequencing quality.
The only difference between DRUG-seq and standard RNA-seq data analysis is the demultiplexing step, which is used to assign sequencing reads to their sample of origin based on the DRUG-seq barcode sequence.
For a thorough description of DRUG-seq data processing, please refer to the DRUG-seq kit data analysis user guide.
The barcode set for your kit is conveniently located on the kit label. Please refer to the label for accurate identification.
For optimal compatibility, ensure that you use the appropriate plate format (e.g., for kits designed for 96 reactions, the 96 well-plate format should be used). This ensures accurate and efficient processing of your samples. If you have any further questions or concerns, please contact our support team for assistance by email or using our live chat tool.
DRUG-seq is used to generate transcriptome-wide gene expression profiles after compound, genetic, or other perturbations. In drug discovery pipelines, these profiles can support hit triage, mechanism-of-action studies, toxicity profiling, dose-response analysis, compound clustering, and AI-ready perturbation dataset generation. Read more.
DRUG-seq is most useful when a screening team needs richer biological context than single-endpoint assays can provide, but standard RNA-seq is too costly or slow for the number of compounds, doses, time points, or replicates being tested.
Yes. DRUG-seq generates transcriptional signatures that can be compared across compounds, controls, doses, and perturbations. Compounds with related mechanisms can show similar gene expression patterns, supporting MoA inference, pathway analysis, and follow-up assay selection.
Yes. DRUG-seq can provide a molecular readout to complement morphological phenotypes from Cell Painting or other high-content imaging assays, helping connect cellular phenotypes to pathway-level transcriptional responses. Read more.
Yes. DRUG-seq can generate structured gene expression matrices across many compounds, concentrations, cell models, and time points, making it useful for predictive modeling, compound similarity analysis, and perturbation-response datasets. In fact, DRUG-seq is an ideal method for generating foundational datasets for AI drug discovery. Read more.
DRUG-seq is optimized for scalable 3′ mRNA counting and gene-level expression profiling. It is not the best choice when the main goal is isoform discovery, fusion detection, alternative splicing, or full-length transcript analysis. For those applications, MERCURIUS™ Total DRUG-seq is a more appropriate method.
* Contact us to inquire about compatibility with other species.
** Find the list of validated cell lines here.
Product
Number of Samples
MERCURIUS™ UDI Expansion module
MERCURIUS™ Cell Lysis Modules
MERCURIUS™ Cell Lysis Modules
MERCURIUS™ Standard Post-Pooling Preparation Module (4 libraries)
Each kit includes 4 UDI pairs. Add the expansion module if you need more unique indexes (total 16 UDI pairs available).
For the first time, 1536-well whole transcriptome profiling brings biologically rich readouts to primary screening and large-scale perturbation dataset generation. In this webinar, Alithea Genomics…
Biomarker discovery using gene expression signatures has transformed pharmaceutical discovery and development, from early hit triage and target validation to toxicology, dose optimization, and patient…
Next-generation AI models for drug discovery are only as powerful as the quality, size, and perturbation-richness of their training data. Yet no publicly available dataset…
As the SOT Annual Meeting and ToxExpo 2026 approaches, we take a pre-conference look at the program through a transcriptomics-focused lens. One overall shift is…