Our BRB-seq service enables industrial and academic scientists to outsource unbiased gene expression profiling at scale, with no trade-off in data quality compared to standard methods.
Discover Sample Datasets
Mammals, Plants, Insects, Fish
The MERCURIUS™ BRB-seq service offers a convenient and scalable solution for transcriptome profiling projects of any size.
Clients send us purified RNA samples, and our team performs rigorous quality control before preparing BRB-seq libraries, sequencing, and performing data pre-processing.
Results are then delivered, including raw sequencing data (fastq files), QC metrics, sequencing and alignment reports, and gene-count matrices for downstream analysis.
During the process, we always keep clients informed at defined checkpoints so that we can decide together how to best proceed to the next steps.

(Nanodrop and Fragment analyzer)
1 week
2 days
(Qubit, Fragment analyzer, shallow sequencing)
1 week
1 week
Sample preparation guidelines for purified RNA (PDF)Raw FASTQ files, sample report file, QC files, and gene count tables
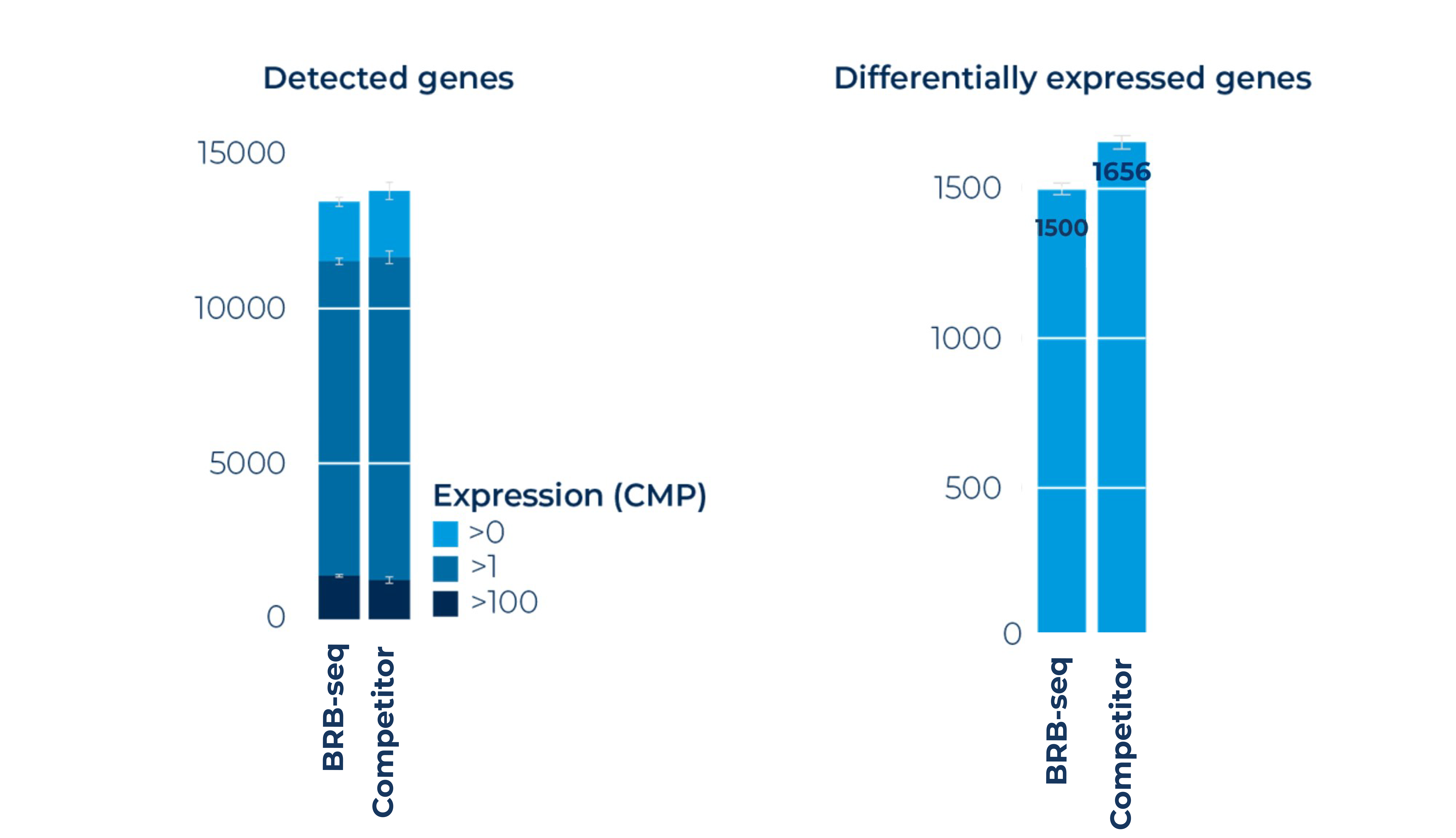
Comparison of gene detection (left) and differential expression (right) between the same samples prepared with MERCURIUS™ BRB-seq or Illumina TruSeq. (Left) Both protocols detect equivalent numbers of genes across all expression thresholds at the same sequencing depth. (Right) Differential expression analysis yields comparable numbers of differentially expressed genes for both technologies. MERCURIUS™ BRB-seq achieves TruSeq-equivalent analytical performance at significantly higher throughput and lower cost per sample.
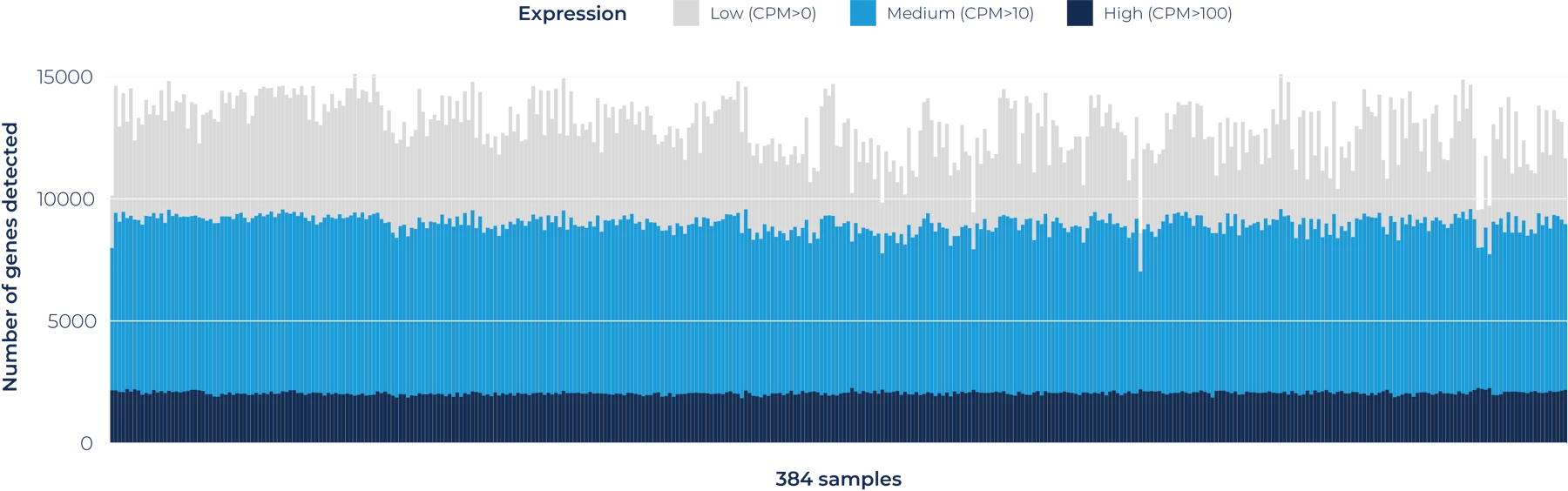
Number of genes detected per sample across a 384-sample BRB-seq run, stratified by expression level. Across all 384 samples, an average of ~13,000 genes is detected when sequenced at a depth of 1.5M reads/sample. The uniformity of gene detection across all samples demonstrates the reproducibility and scalability of MERCURIUS™ BRB-seq for high-throughput transcriptomic profiling.
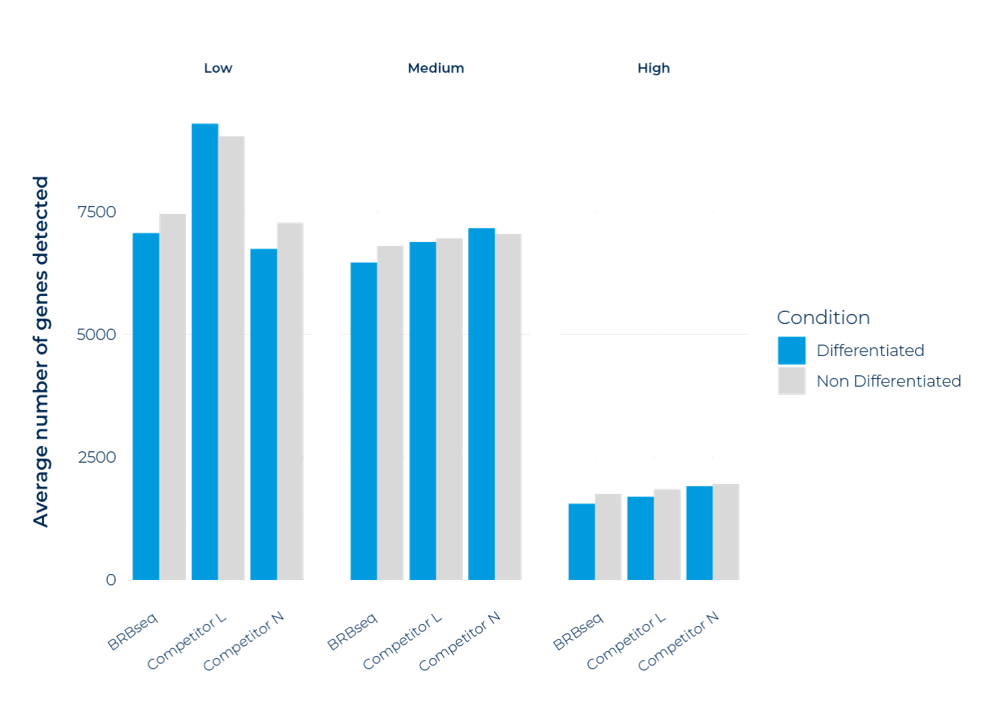
Average number of genes detected per expression category in non-differentiated (ND) and differentiated (D) adipose stromal population cells, comparing MERCURIUS™ BRB-seq against two commercial competitor kits. Genes are stratified into lowly expressed (0 < CPM < 10), mid-expressed (10 < CPM < 100), and highly expressed (CPM > 100) categories. All replicates were prepared from the same RNA sample and sequenced at a uniform depth of 3 million reads per sample. MERCURIUS™ BRB-seq performs comparably to both competitors across all expression levels and cell states.
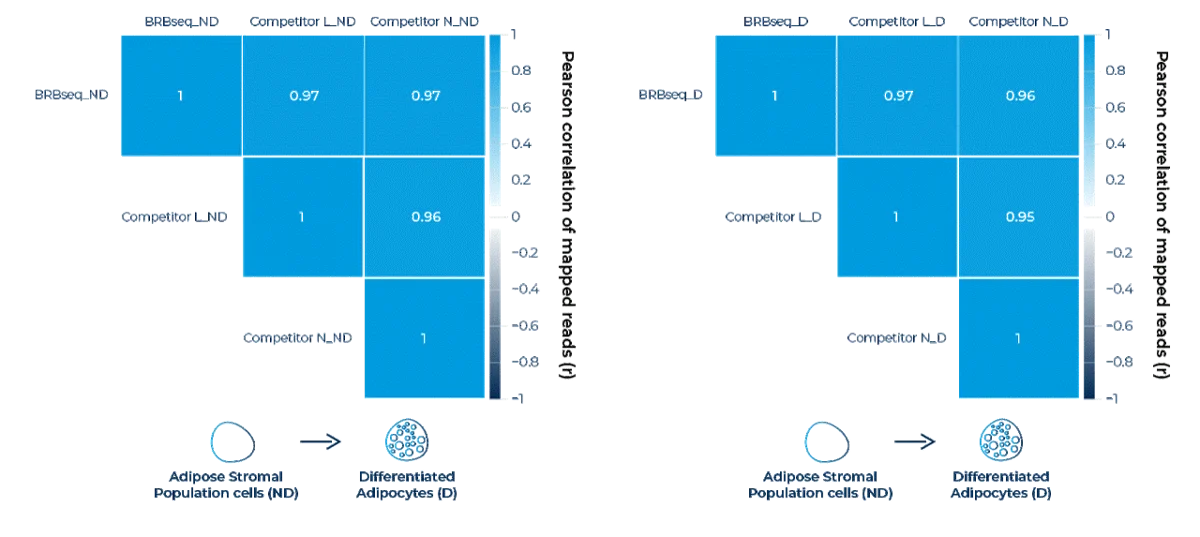
Transcriptome-wide Pearson correlation matrices of mapped reads comparing MERCURIUS™ BRB-seq against Competitor L and Competitor N in non-differentiated adipose stromal Population cells (ND, left) and differentiated adipocytes (D, right). MERCURIUS™ BRB-seq achieves Pearson correlation coefficients of r ≥ 0.97 against both competitors in both cell states, comparable to the inter-competitor correlation (r = 0.95–0.96), demonstrating that MERCURIUS™ BRB-seq produces highly concordant gene expression profiles regardless of biological condition.
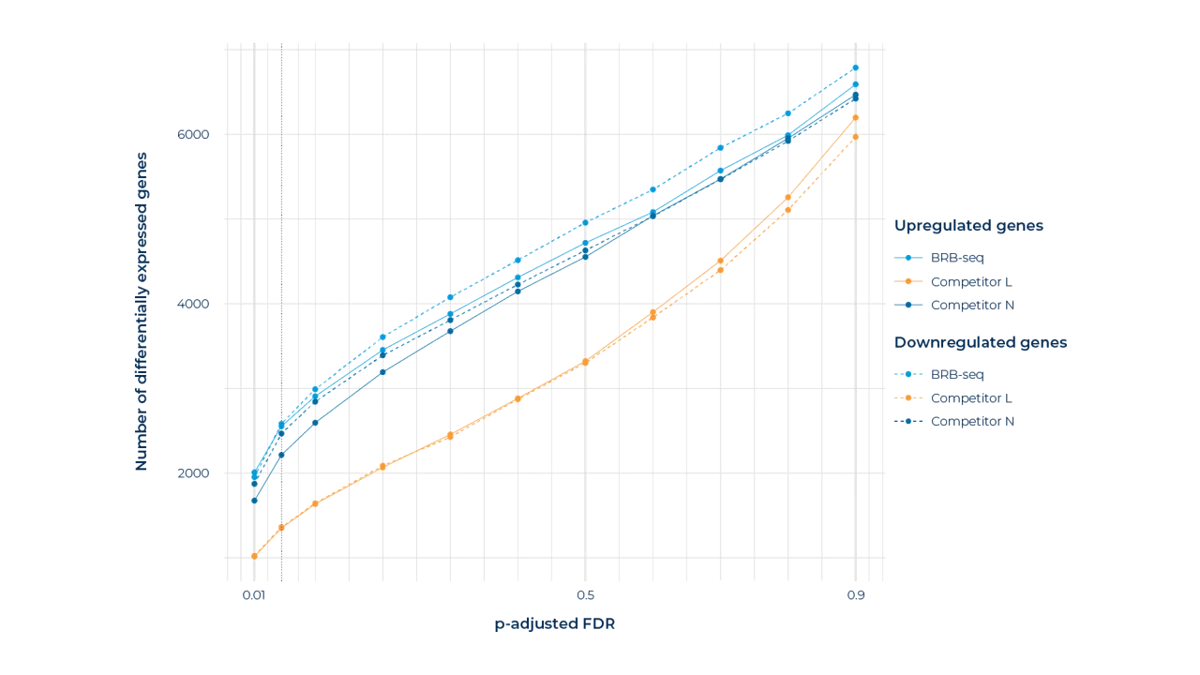
Number of up and downregulated differentially expressed genes (DEGs) detected by MERCURIUS™ BRB-seq, Competitor L, and Competitor N as a function of FDR-adjusted p-value threshold, comparing non-differentiated adipose stromal population cells to differentiated adipocytes at 3M reads/sample on the same initial RNA. MERCURIUS™ BRB-seq consistently identifies more DEGs than both competitors across the full range of significance thresholds, suggesting greater sensitivity for capturing the full transcriptional response to differentiation.
To access the deep-sequenced dataset (9.5M reads per sample), contact us.
To access the deep-sequenced dataset (8.9M reads per sample), contact us.
To access the deep-sequenced dataset (5.7M reads per sample), contact us.
To access the deep-sequenced dataset (6.3M reads per sample), contact us.
To access the deep-sequenced dataset (9M reads per sample), contact us.
To have access to the deep-sequenced dataset (3M reads per sample) contact us.
To have access to the deep-sequenced dataset (9M reads per sample) contact us.
To have access to the deep-sequenced dataset (3M reads per sample) contact us.
To have access to the deep-sequenced dataset (4M reads per sample) contact us.
To have access to the deep-sequenced dataset (5.3M reads per sample) contact us.
To have access to the deep-sequenced dataset (4.7M reads per sample) contact us.
To guarantee high quality data, we recommend that each sample contains at least 12ng/μl of total RNA in at least 15μl.
In addition to total RNA amount, it is important that the samples contain RNA of high integrity (RIN >= 6) and are devoid of contaminants (Nanodrop A260/A230 between 1.8 and 2.2).
BRB-seq is 3’-end RNA sequencing method and, as such, requires significantly less sequencing compared to standard full-length RNA-seq in order to reach accurate gene quantification. We therefore recommend sequencing 4 to 5 million reads per sample, enabling reliable, unbiased detection of over 18,000 genes.
As part of our standard service pipeline, we align the generated data to the genome of choice, provide a detailed report on the alignment and gene counting statistics and, finally, provide ready-to-use gene count matrices for downstream analysis.
You can either book a call with our experts to discuss your project or submit your experimental details via our contact form so we can review your design and requirements.
Based on the goals, sample types, and scale of your study, we may recommend starting with a pilot project to optimise conditions and de-risk a larger screen.
If you’re interested in implementing the technology in your own lab instead, you can explore our MERCURIUS™ BRB-seq kits on the dedicated kits page.
Determining the most suitable transcriptomic technology to drive your large-scale compound screen, clinical study, or to assess a panel of genetic perturbations can be a…
High-throughput’ in sequencing refers to the amount of DNA molecules read at the same time. Technologies are now capable of sequencing many fragments of DNA…
With a growing number of published 3’ mRNA-seq methods now available, researchers have more choices than ever for high-throughput and cost-effective transcriptomic screening. While broadly…