
Revolutionizing Single-Cell RNA Sequencing with FLASH-seq
The field of genomics is advancing rapidly, and one of the most transformative tools has been single-cell RNA sequencing (scRNA-seq). This technology enables researchers to explore cellular diversity and gene expression at an unmatched level of detail, providing insights into how individual cells function within tissues and organs(1,2,3). Methods in the scRNA-seq field can be broadly divided into two categories. On one side, microfluidics and combinatorial indexing methods typically assay thousands of cells in parallel, capturing only the 3’ or 5’ end of the transcripts and display low gene detection (= sensitivity). These include approaches from 10x Genomics or Parse Biosciences. On the other side, plate-based scRNA-seq methods have lower throughput but high sensitivity and provide information across the entire body of a gene (= full-length). The most famous plate-based method is Smart-seq2(4).
Among the latest breakthroughs in scRNA-seq is FLASH-seq(5), a cutting-edge plate-based method designed to enhance both the sensitivity and efficiency of single-cell RNA sequencing. In this blog, we explore how FLASH-seq is poised to reshape the landscape of single-cell transcriptomics.
The Need for Better scRNA-seq Methods
While traditional scRNA-seq methods like 10x Genomics have revolutionized our understanding of cellular heterogeneity, they come with notable limitations. These include slow processing times, dependency on specific instruments, inability to process limited or rare cell population, biased coverage (3’ or 5’-only) and low sensitivity—leading to incomplete data on gene expression. As the field continues to grow, there has been a pressing need for more efficient and sensitive methods to meet the increasing demands of research and clinical applications.
This is where FLASH-seq steps in, offering a significant leap forward by addressing the key limitations of earlier techniques.
Introducing FLASH-seq
FLASH-seq represents a major advancement in scRNA-seq technology. It brings several key improvements over its predecessors:
- Enhanced Sensitivity: FLASH-seq excels at detecting more genes, particularly protein-coding and longer genes. This increased sensitivity allows for a deeper and more comprehensive gene expression analysis, giving researchers a clearer picture of cellular processes (Figure 1). Compared to 10x Genomics, at the same sequencing depth, FLASH-seq detects up-to 3-times more genes.
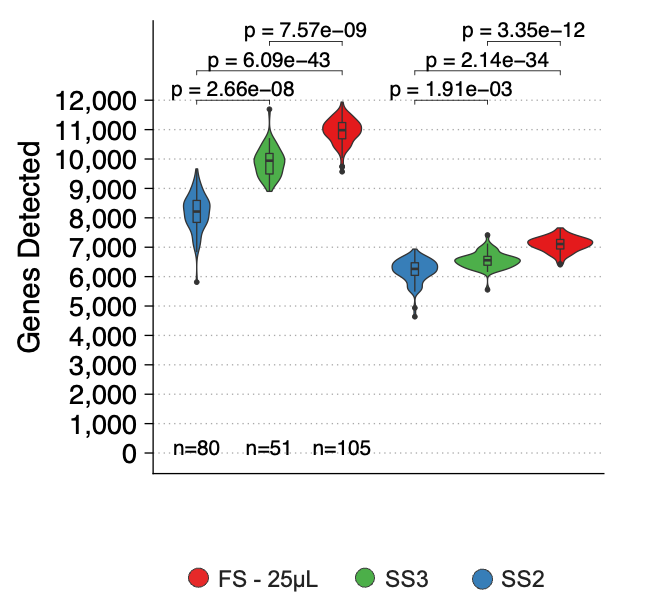
Figure 1- Number of genes detected in HEK293T cells processed with SS2 (n = 80), SS3 (n = 51) and FS (n = 105) at two read thresholds, with reads downsampled to 500,000 (= 500K) raw reads. Taken from (5).
- Faster Processing: Traditional scRNA-seq methods can be slow and resource-intensive, but FLASH-seq dramatically reduces the time required before sequencing. The entire protocol can be completed in just 7.5 hours with the regular protocol and 4.5 hours with the “low-amplification” version, making it ideal for high-throughput projects on rare cells that require a high sensitivity. Moreover, the reduced number of steps simplifies integration into automated systems (Figure 2).
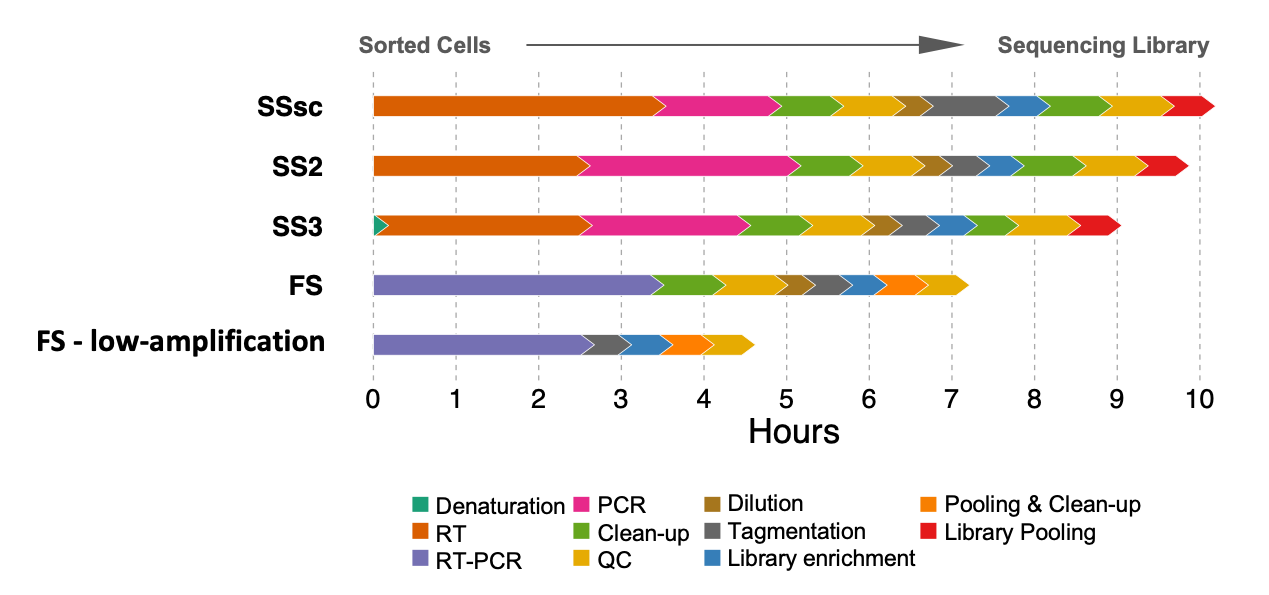
Figure 2 – Estimated protocol duration to process a 96-well plate of HEK293T cells for the full-length scRNA-seq protocols used in this study. Steps are color-coded. QCs include concentration and size distribution measurements. SSsc, SMART-Seq Single Cell Kit (Takara). Updated from (5).
- Minimized Strand-Invasion Artifacts: A common problem with older techniques is the occurrence of strand-invasion artifacts, which can skew results. FLASH-seq uses a novel approach to minimize these errors, resulting in more accurate and reliable data.
- Improved Gene-Body Coverage: FLASH-seq provides full-length gene-body coverage, enabling the detection of alternative splicing or fusion events and capturing regulatory mechanisms that are not accessible via 3’ or 5’ biased methods, such as 10x Genomics (Figure 3). Moreover, even single nucleotide polymorph can be called at the single-cell level.
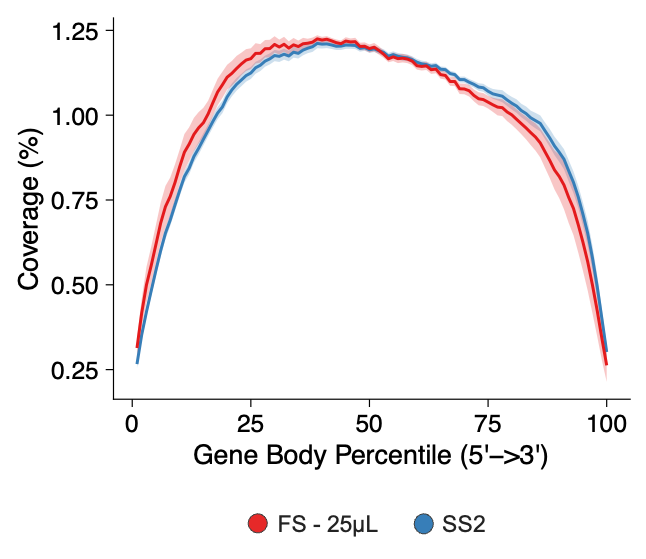
Figure 3 – Mean ± standard deviation (s.d.) gene-body coverage in HEK293T cells. Taken from (5).
The Technology Behind FLASH-seq
The success of FLASH-seq lies in its streamlined and innovative approach to sequencing. The process involves several key steps:
- FACS sorting: Individual cells are FACS sorted in a multi-well plate (96 or 384).
- Cell Lysis and RNA Capture: Upon contact with the lysis buffer, cells are broken open, releasing their RNA, which is then captured using oligo(dT) primers that bind to the poly(A) tails of mRNA molecules.
- Reverse Transcription and cDNA amplification: The captured RNA is reverse transcribed into complementary DNA (cDNA) and amplified within a single RT-PCR reaction.
- Amplification and Library Preparation: cDNA molecules are fragmented and PCR amplified using cell-specific indexes. FLASH-seq integrates a “low-amplification” version of the protocol where cDNA molecules can be directly fragmented without any prior purification or dilution.
- Sequencing and Data Analysis: High-throughput sequencing platforms are used to analyze the prepared libraries, yielding data that is then processed to quantify gene expression and uncover alternative splicing events.
Applications and Impact of FLASH-seq
The potential of FLASH-seq spans across numerous fields of biomedical research. It is especially useful for studying rare cells, that otherwise would not be possible. Here are just a few areas where it could make a significant impact:
- Cancer Research: Cancer often relapses after treatment due to the presence of a small number of resistant cells. FLASH-seq offers the resolution and sensitivity needed to identify new therapeutic targets by revealing the heterogeneity within these rare cell populations.
- Developmental Biology: By mapping gene expression trajectories across individual cells during early development, FLASH-seq allows researchers to study how cells differentiate and form tissues, helping to unravel the complex processes that drive organismal development.
- Neuroscience: The brain consists of a vast array of cell types, each with distinct roles. FLASH-seq can map the transcriptomic landscape of these cells, contributing to a deeper understanding of neuronal functions and the molecular mechanisms underlying neurological disorders. The protocol has been successfully used to work on rare, sorted cell types in the retina or brain as well as sub-cellular content obtained through PATCH-seq experiments(6).
- Immunology: The immune system’s complexity lies in the diverse cell types it comprises. FLASH-seq can track gene expression in immune cells, offering insights into how the immune system responds to infections, autoimmune disorders, and other diseases. Moreover, TCR chains and CDR3 variable regions can be inferred at the single-cell level from FLASH-seq data without any additional processing(5).
- Sub-cellular content: The sensitivity of FLASH-seq makes it efficient even with sub-cellular amounts of RNA (<10pg) such as exosomes preparations or biopsies of individual cells (PATCH-seq).
Future Directions and Challenges
Although FLASH-seq offers a transformative approach to scRNA-seq, challenges remain. One limitation is its reliance on FACS-sorted cells, which could restrict its use with certain sample types. Additionally, the low-amplification version of the protocol requires some titration to estimate the right number of PCR cycles to use during cDNA amplification. Finally, FLASH-seq appears more prone to capture MT-RNA than other methods. However, as technology continues to evolve, it’s likely that FLASH-seq will become even more versatile and powerful.
Conclusion
In summary, FLASH-seq is a groundbreaking advancement in single-cell RNA sequencing, offering a combination of speed, sensitivity, and accuracy that surpasses traditional methods. Its ability to detect more genes, provides better full-length coverage, and reduces sequencing errors making it a game-changer for researchers delving into the complexities of gene expression and cellular diversity.
As FLASH-seq and similar technologies continue to evolve, they will undoubtedly accelerate discoveries in fields ranging from cancer research to neuroscience, paving the way for new therapeutic strategies and a deeper understanding of biology at the single-cell level.
References
- Picelli, S. et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 10, 1096–1098 (2013).
- Deng, Q., Ramskold, D., Reinius, B. & Sandberg, R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343, 193–196 (2014).
- Byrne, A. et al. Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat. Commun. 8, 16027 (2017).
- Picelli, S., Faridani, O., Björklund, Å. et al. Full-length RNA-seq from single cells using Smart-seq2.Nat Protoc 9, 171–181 (2014). https://doi.org/10.1038/nprot.2014.006
- Hahaut, V., Pavlinic, D., Carbone, W. et al. Fast and highly sensitive full-length single-cell RNA sequencing using FLASH-seq. Nat Biotechnol 40, 1447–1451 (2022). https://doi.org/10.1038/s41587-022-01312-3
- Marx, V. Patch-seq takes neuroscience to a multimodal place. Nat Methods 19, 1340–1344 (2022). https://doi.org/10.1038/s41592-022-01662-5