
3’ mRNA-seq is a quantitative, genome-wide transcriptomic technique based on the barcoding of the 3’ untranslated region (UTR) of mRNA molecules. Unlike standard bulk RNA-seq, where short sequencing reads are generated along the entire length of mRNA transcripts, only the 3’ end of polyadenylated RNAs are sequenced in 3’ mRNA-seq. This approach results in a need for fewer reads to quantify the expression of a gene and reduces the sequencing depth required per sample while providing robust and reliable transcriptome-wide read-outs of gene expression levels comparable to full-length RNA-seq methods (Alpern et al., 2019; Ma et al., 2019).
Sample barcoding and the reduced per-sample sequencing depth also allow higher levels of sample multiplexing per experiment and lower the cost of transcriptome sequencing compared to full-length RNA-seq methods. These factors are crucial for large-scale, ultra-high-throughput gene expression studies or studies assessing differential gene expression between different experimental conditions or cell types.
Some 3’ mRNA-seq technologies, like Bulk RNA Barcoding and Sequencing (BRB-seq) commercialized by as MERCURIUS™ BRB-seq, further streamline the library preparation process by pooling up to 384 samples very early in the workflow for a cost per sample tantamount to profiling four individual genes using conventional qRT-PCR, in a workflow requiring less than two and a half hours hands-on time. An increasing number of 3’ mRNA-seq techniques, like BRB-seq, also include unique molecular identifiers (UMIs) in sample barcodes to uniquely label each mRNA molecule and to distinguish between original mRNA transcripts and duplicates that result from PCR amplification.
History
The sample barcoding approach used in 3’ mRNA-seq was first established in the field of single-cell transcriptomics, where sample and mRNA barcoding allowed hundreds to thousands of single cells to be multiplexed in one experiment (Ziegenhain et al., 2017). Single-cell RNA profiling technologies like CEL-seq2, SCRB-seq, and STRT-seq also allowed the pooling of large sets of samples into one unique sequencing library early in the protocol. This early multiplexing reduced overall experimental costs and hands-on time while boosting throughput and is a fundamental aspect of some 3’ mRNA-seq technologies.
Method
While numerous different 3’ mRNA-seq methods are available, their fundamental principles are generally similar (Fig. 1).
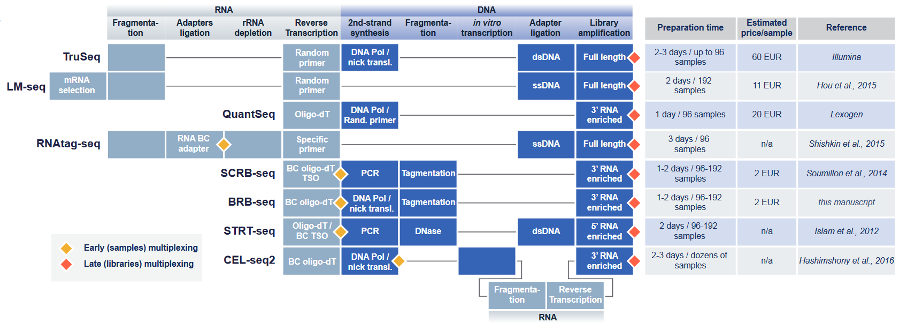
Figure 1. State-of-the-art bulk and single-cell RNA-seq approaches allow different levels of sample multiplexing. Every box corresponds to a standardized step of the protocol. Diamonds represent the multiplexing steps: early (yellow) or late (red). The right table shows the throughput of each protocol and the estimated library preparation cost per sample in Euros. Legend: BC, barcoded; TSO, template switch oligo; ssDNA: single-stranded DNA; dsDNA: double-stranded DNA. (Figure from Alpern et al., 2019).
Each method relies on an initial reverse transcription step in which mRNAs are labeled with sample barcodes. Reverse transcription can be performed with oligo dT primers, barcoded oligo dT primers, and template-switching oligos. In contrast, bulk RNA-seq library preparation methods like Illumina TruSeq mRNA Stranded kits use random priming of pre-fragmented RNA for reverse transcription to ensure reads are generated along the entire length of mRNA transcripts.
Second-strand synthesis is then performed in each method by DNA polymerase 1 nick translation or PCR, resulting in double-stranded complementary DNA (cDNA). This is followed by a process called tagmentation, in which double-stranded cDNA is fragmented and tagged using Tn5 transposase, which cleaves the cDNA and ligates adaptors for library amplification. Some methods use random primers for this stage.
Library indexing and PCR amplification then take place, resulting in libraries enriched for the 3’ untranslated region of mRNAs and suitable for short-read sequencing on Illumina or MGI sequencing instruments.
Advantages of 3’ mRNA-seq
Cost
3’ mRNA-seq methods are generally cheaper per sample than standard bulk RNA-seq methods. This is because of the lower sequencing depth required as only the 3’ end of mRNA molecules are sequenced instead of the whole length of entire transcripts. Read depths of between one million and five million reads are recommended in commercialized 3’ mRNA-seq protocols and are suitable for detecting the majority of highly expressed genes. For methods where samples are pooled early in the workflow, consumable use is also reduced. For instance, BRB-seq is up to 25 times cheaper than Illumina TruSeq stranded mRNA library preparations, with a cost equivalent to assessing four genes by RT-qPCR.
Sample throughput
3’ mRNA-seq methods have a higher sample throughput than standard RNA-seq approaches. Different options allow up to 384 samples to be barcoded and multiplexed at different stages in the library preparation protocol, followed by short-read sequencing. 3’ mRNA-seq methods are performed in 96 or 384 well plates and are suitable for automated pipelines.
Ease of data analysis
As only the 3’ end of mRNA molecules are sequenced, read counts do not need to be normalized for transcript length as in standard RNA-seq methods. Similarly, the data size per sample is lower than that of standard RNA-seq due to the lower sequencing depth required. This simplifies data analysis with options available for rapid cloud-based analyses.
Suitable for degraded RNA
3’ mRNA-seq is suitable for high-quality and degraded RNA with RIN <6 (1). It is largely insensitive to RNA degradation because only the 3’ region of mRNA transcripts are prepared for sequencing.
Limitations of 3’ mRNA-seq
3’ mRNA-seq methods do not allow for the analysis of full-length transcripts, splice variants, fusion genes, or RNA editing.
3′ mRNA-seq by Alithea Genomics
Various commercialized 3’ mRNA-seq methods are available to researchers as kits or as a whole service. Each technology has different multiplexing capabilities, input RNA quality and quantity requirements, and overall performance.
Alithea Genomics has commercialized BRB-seq 3’ mRNA-seq technology, with different options available under the MERCURIUS™ family of technologies (Fig. 2). Samples are pooled early in the workflow and are processed simultaneously, reducing hands-on time, cost, and consumable use. Kits compatible with Illumina, Element Bioscience and MGI sequencers, alongside full-service options, are available and are suitable for up to 384 samples per kit.
MERCURIUS™ DRUG-seq from Alithea Genomics is a modified version of the MERCURIUS™ BRB-seq 3’ mRNA-seq technology. It has the added advantage that it is RNA-extraction-free and can be performed directly on cell and organoid lysates, requiring no prior RNA isolation. This makes it suitable for ultra-high-throughput transcriptomic experiments and removes the costly and time-consuming RNA-extraction stages.
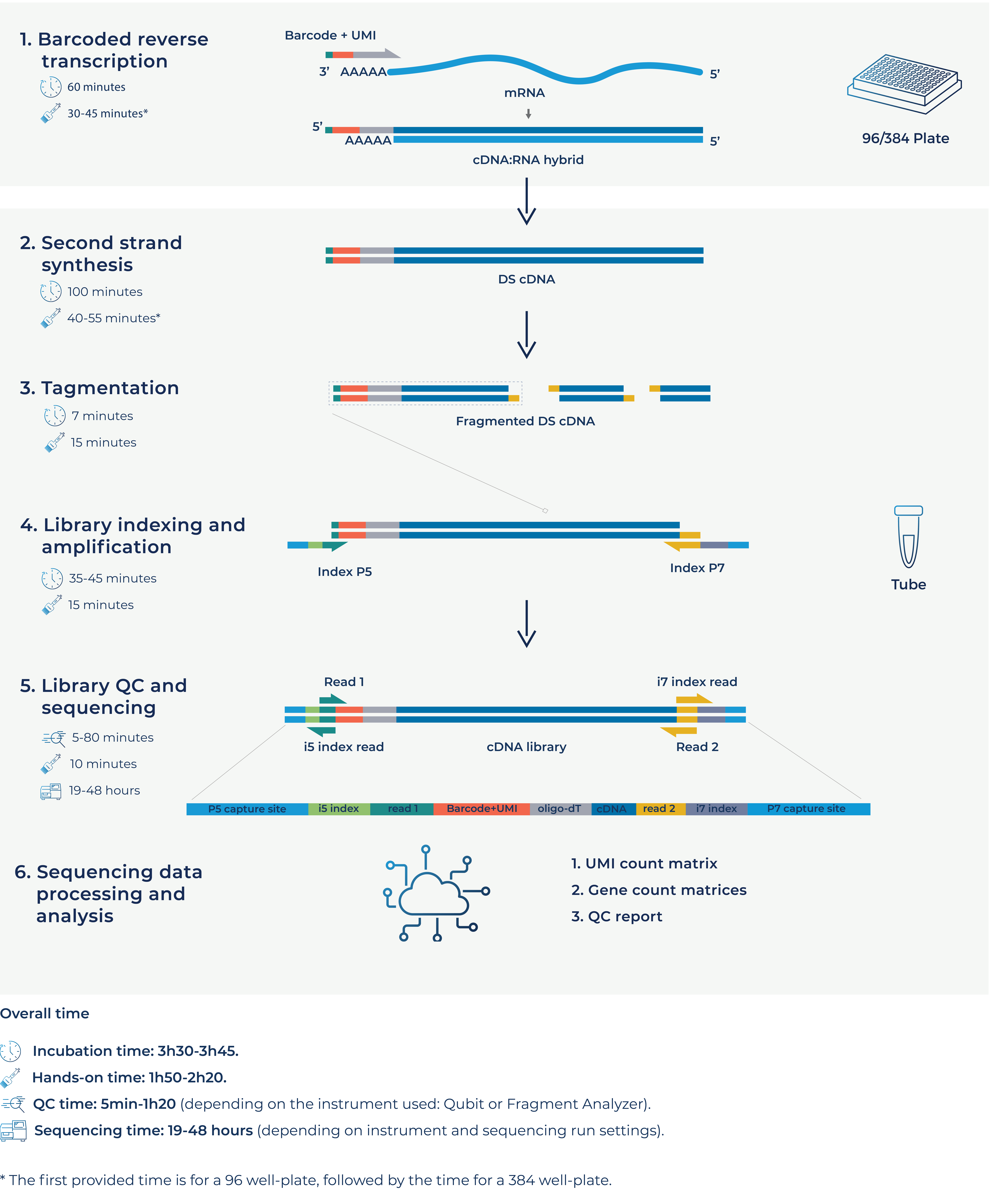
Figure 2. Schematic overview of the MERCURIUS™ BRB-seq workflow where up to 384 samples can be barcoded and multiplexed per kit.
Contact us for more information about our technologies, kits, or services.
References
- Alpern, Daniel, Vincent Gardeux, Julie Russeil, Bastien Mangeat, Antonio CA Meireles-Filho, Romane Breysse, David Hacker, and Bart Deplancke. “BRB-seq: ultra-affordable high-throughput transcriptomics enabled by bulk RNA barcoding and sequencing.” Genome Biology 20 (2019): 1-15.
- Ma, Feiyang, Brie K. Fuqua, Yehudit Hasin, Clara Yukhtman, Chris D. Vulpe, Aldons J. Lusis, and Matteo Pellegrini. “A comparison between whole transcript and 3’RNA sequencing methods using Kapa and Lexogen library preparation methods.” BMC Genomics 20 (2019): 1-12. https://link.springer.com/article/10.1186/s12864-018-5393-3
-
- Ziegenhain, Christoph, Beate Vieth, Swati Parekh, Björn Reinius, Amy Guillaumet-Adkins, Martha Smets, Heinrich Leonhardt, Holger Heyn, Ines Hellmann, and Wolfgang Enard. “Comparative analysis of single-cell RNA sequencing methods.”
Molecular Cell
- 65, no. 4 (2017): 631-643.
https://www.cell.com/molecular-cell/pdf/S1097-2765(17)30049-7.pdf