Full-length and total RNA-seq combined with massive sample multiplexing.
Our MERCURIUS™ Total BRB-seq service is a convenient and scalable solution for projects of any size, enabling transcriptome-wide detection of full-length coding and non-coding transcripts.
As part of the Total BRB-seq service, users simply deliver their 96-well plates containing frozen purified RNA to us in Switzerland or the United States.
Upon receipt of the plates, our team prepares the libraries, then sequences to your desired read depth per sample, and then performs data pre-processing.
We return results, including raw fastq files, sequencing and alignment reports, and gene count matrices suitable for downstream differential expression analyses, once the data meet our rigorous quality control criteria.
During the process, we always keep clients informed at defined checkpoints so we can decide together how best to proceed to the next steps.

(Nanodrop and Fragment analyzer)
1 week
2 days
(Qubit, Fragment analyzer, shallow sequencing)
1 week
1 week
1 week
Raw FASTQ files, sample report file, QC files, and gene count tables
Saturation curves indicating the number of detected genes as a function of sequencing depth in Huh7 cells. MERCURIUS™ Full-Length BRB-seq and Total BRB-seq perform comparably to NEBNext® Ultra™ II mRNA and Total RNA preparations, respectively, demonstrating equivalent sensitivity at a fraction of the library preparation cost and effort. MERCURIUS™ BRB-seq is a 3’ mRNA library preparation technology, hence the lower overall gene detection versus full-length methods.

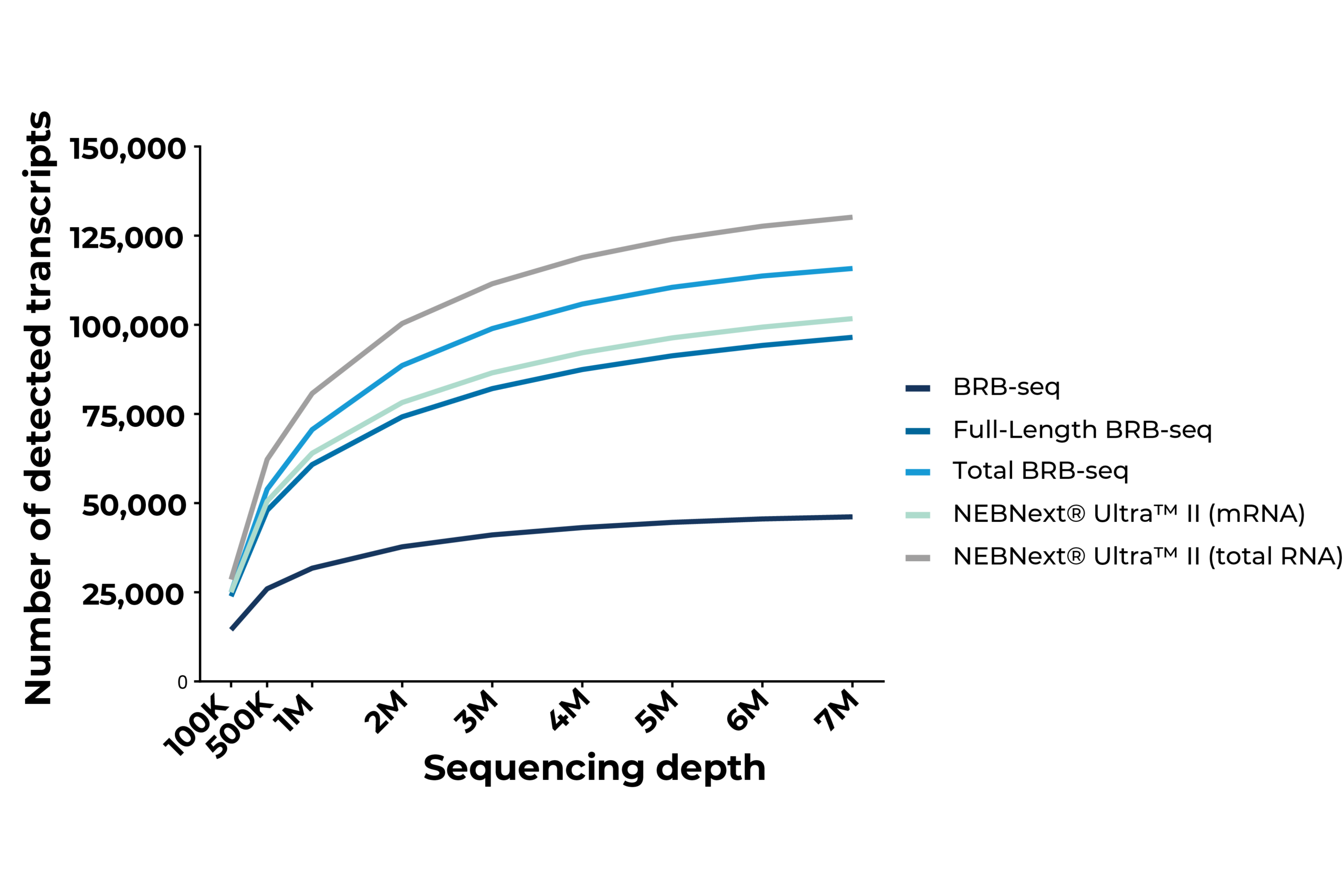
Saturation curves indicating the number of detected transcripts as a function of sequencing depth in Huh7 cells. MERCURIUS™ Full-Length BRB-seq and Total BRB-seq perform comparably to NEBNext® Ultra™ II mRNA and Total RNA preparations, respectively, demonstrating equivalent sensitivity at a fraction of the library preparation cost and effort. MERCURIUS™ BRB-seq specifically detects protein-coding transcripts, hence the lower overall transcript detection compared to full-length methods.
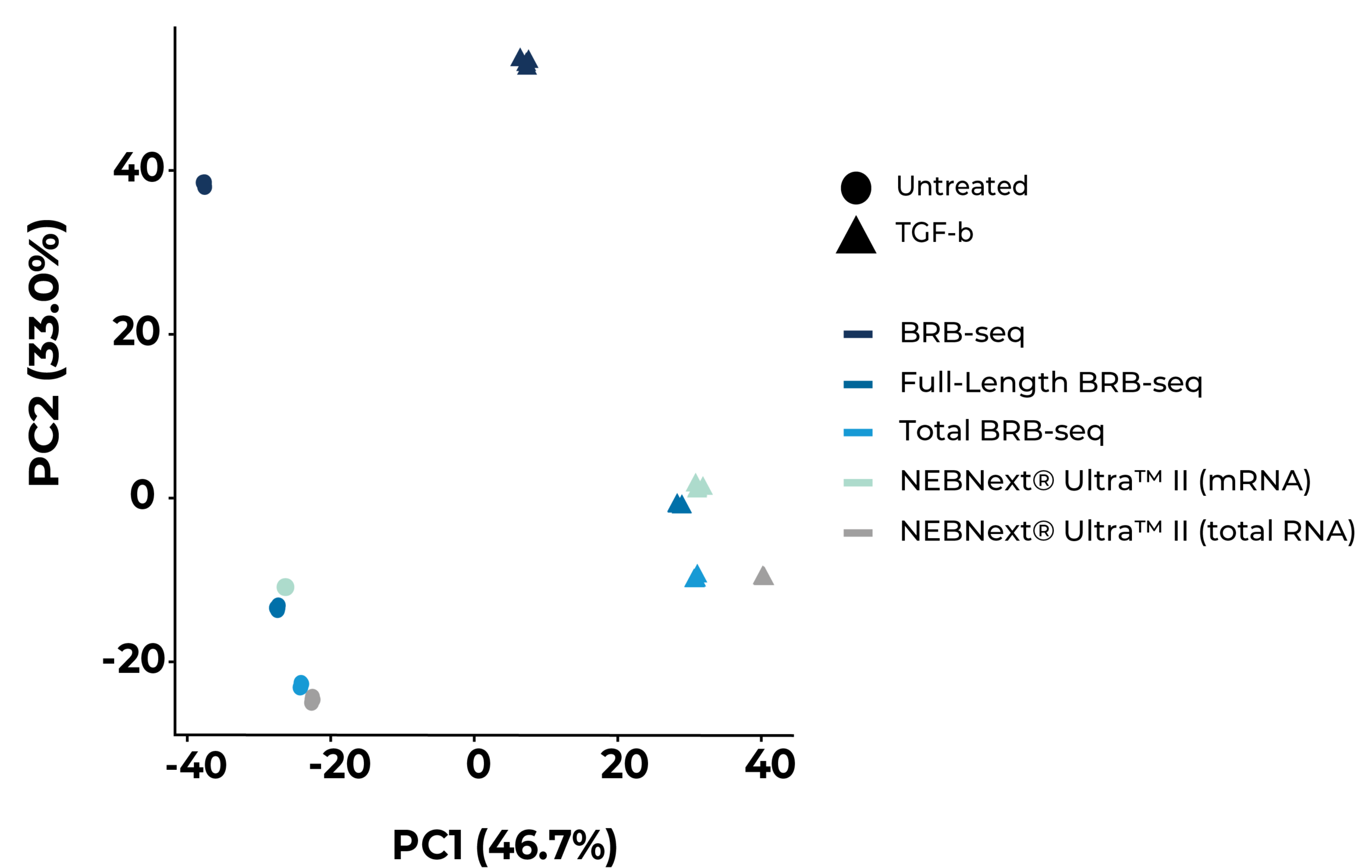
Principal component analysis (PCA) of gene expression profiles generated by the specified library preparation methods across untreated (circles) and TGF-β-treated (triangles) Huh7 samples. Samples cluster primarily by library type along PC2 and by treatment condition along PC1. All methods robustly capture the expression response to treatment. MERCURIUS™ Full-Length BRB-seq and Total BRB-seq cluster closely with their NEBNext® Ultra™ II counterparts, demonstrating equivalent transcriptomic profiles. MERCURIUS™ BRB-seq clusters separately, consistent with its 3′-end capture design.
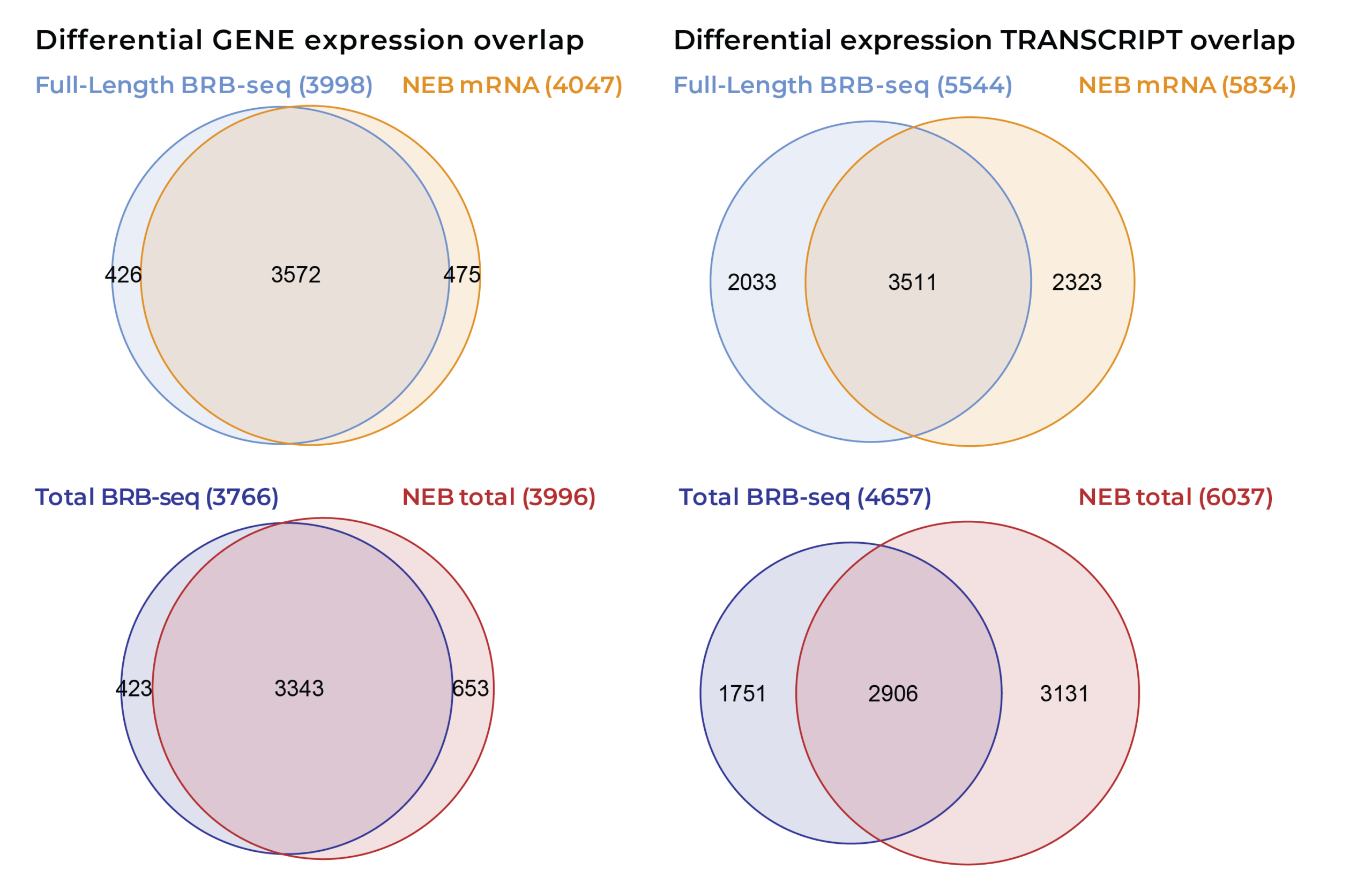
Venn diagrams showing the overlap in differentially expressed (DE) genes (left) and transcripts (right) identified by MERCURIUS™ Full Length BRB-seq and NEBNext® Ultra™ II for mRNA libraries (top) and total RNA libraries (bottom). Numbers indicate DE features unique to each method or shared between both. At the gene level, MERCURIUS™ Full Length BRB-seq and NEBNext® Ultra™ II share over 88% of DE genes for mRNA and 84% for total RNA preparations, demonstrating strong concordance in differential expression calls across both library types. The overlap is smaller but still strong for DE transcripts across technologies.
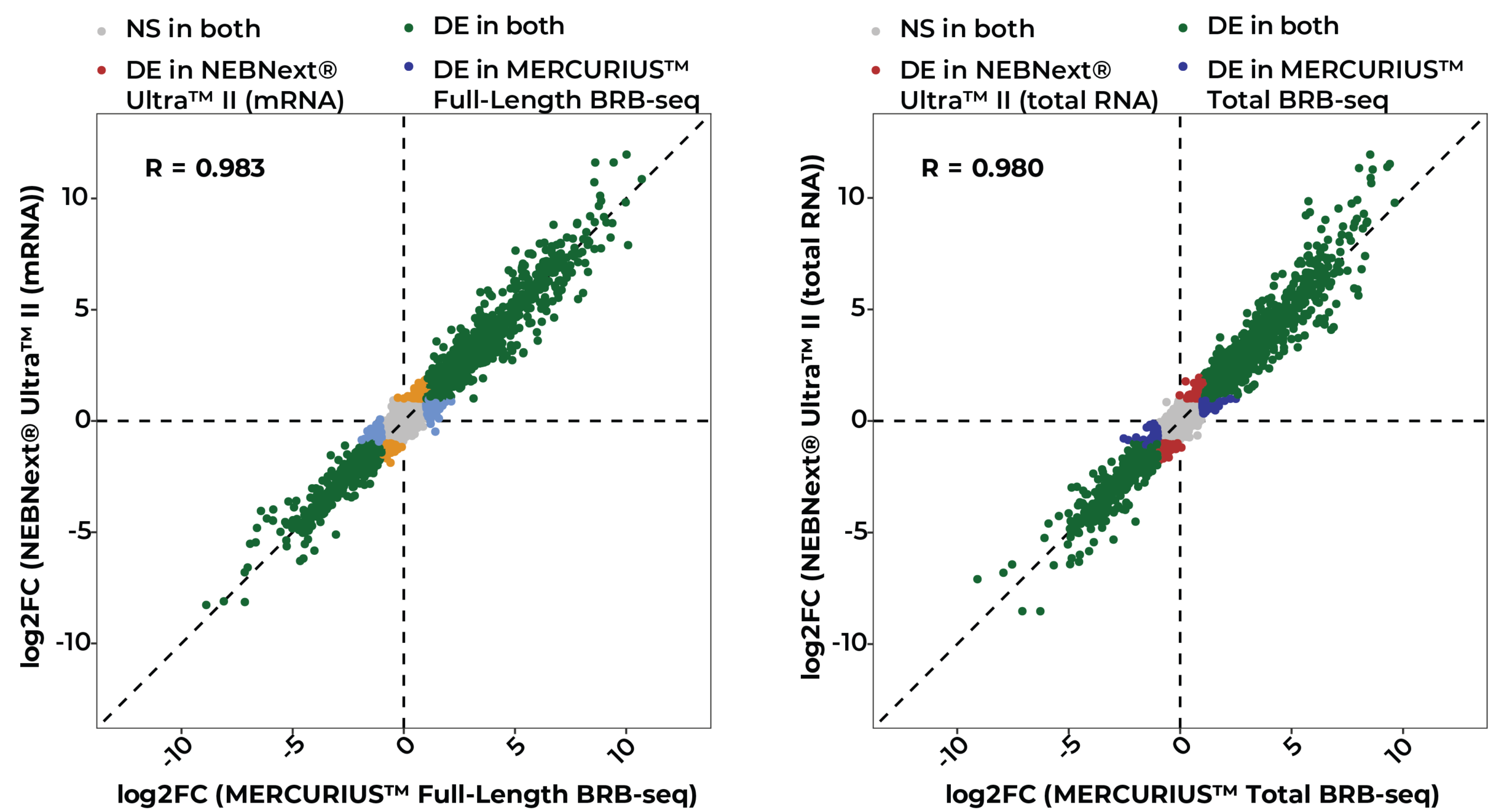
Log2 fold-change correlation between MERCURIUS™ Full-Length BRB-seq and NEBNext® Ultra™ II for mRNA (left, R = 0.983) and total RNA (right, R = 0.980) protocols, across intersecting gene sets of 13,364 and 13,237 genes, respectively. Each point represents a gene, colored by its differential expression (DE) or non-significant (NS) status after treatment of Huh7 cells with human TGF-β versus untreated cells. The dashed diagonal represents perfect correlation. The concordance of DE fold changes across both RNA input types demonstrates that MERCURIUS™ Full-Length and Total BRB-seq reliably recapitulate the results of gold-standard library preparation.
To generate high-quality sequencing data, we recommend between 100pg to 100ng total purified RNA per sample.
In addition to total RNA amount, it is important that the samples contain RNA of high integrity (RIN > 7) and are devoid of contaminants (Nanodrop A260/A230 > 1.5).
Total BRB-seq provides comprehensive coverage of the full-length RNA transcripts.
We, therefore, normally recommend sequencing 12-20 million reads for each sample, which enables the reliable and unbiased detection of over 20,000 genes.
You can either book a call with our experts to discuss your project or submit your experimental details via our contact form so we can review your design and requirements.
Based on the goals, sample types, and scale of your study, we may recommend starting with a pilot project to optimise conditions and de-risk a larger screen.
If you’re interested in implementing the technology in your own lab instead, you can explore our MERCURIUS™ Total BRB-seq kits on the dedicated kits page.
Determining the most suitable transcriptomic technology to drive your large-scale compound screen, clinical study, or to assess a panel of genetic perturbations can be a…
High-throughput’ in sequencing refers to the amount of DNA molecules read at the same time. Technologies are now capable of sequencing many fragments of DNA…
With a growing number of published 3’ mRNA-seq methods now available, researchers have more choices than ever for high-throughput and cost-effective transcriptomic screening. While broadly…