
Next-generation AI models for drug discovery are only as powerful as the quality, size, and perturbation-richness of their training data. Yet no publicly available dataset provides the scale, breadth, or standardization required to robustly train AI models, and traditional sample-by-sample RNA-seq methods weren’t designed for industrial-scale, high-throughput transcriptomic perturbation screens.
As a result, the pharmaceutical industry struggled, until recently, to generate transcriptomic data across thousands of compounds, doses, and time points. High-throughput transcriptomics now overcomes this data-generation bottleneck and provides the foundation for bespoke AI models to detect biological relationships, predict how cells respond to perturbations, and identify novel targets, biomarkers, and early toxicity, while boosting pipeline efficiency.
Read on to find out how high-throughput transcriptomic technologies like MERCURIUS™ DRUG-seq now help pharma generate standardized 3’ or full-length total RNA transcriptomic datasets from 2D cellular or 3D spheroid samples for thousands of perturbations in a single screen to train more robust AI models and improve both the efficacy and effectiveness of AI-aided drug discovery.
Transcriptomics for AI: Key Takeaways
- AI models need millions of perturbation profiles to train effectively, but public datasets lack the scale, consistency, metadata completeness, and standardization required.
- Traditional RNA-seq can’t generate AI-ready datasets at scale because of excessive cost, time, automation difficulties, and data variability.
- High-throughput transcriptomics, such as MERCURIUS™ DRUG-seq, overcomes the data-generation bottleneck, making it feasible to profile tens of thousands of perturbations in a single experiment.
- Proprietary datasets create a competitive advantage because bespoke training data can’t be replicated or accessed by competitors.
How AI Helps Drug Discovery and Development
Developing a new drug costs an estimated US$2.6 billion and takes approximately 12 to 15 years to complete (1). With a proposed chemical space of between 1060-100 for pharma to explore, discovering a clinically relevant compound is highly complex (2). Even when high-throughput screens do find active compounds, they must undergo rigorous assessments of efficacy, safety, and quality before regulatory approval.
As a result, the desire to accelerate drug discovery while reducing overall costs is driving the increasingly widespread adoption of AI and ML in the pharmaceutical industry. In line with this uptake, the ability of AI models to simultaneously process, understand, and discover novel information from vast amounts of data is already improving rational compound discovery and development compared to traditional approaches.
AI has identified novel disease biomarkers, uncovered potential drug targets, used transcriptomic data to detect early safety concerns missed by traditional toxicology approaches, found drug repurposing opportunities, and determined the efficacy of drug candidates, among a growing number of other use cases (3-7).
How to Train AI Models for Drug Discovery and Development
The choices made at each stage when training an AI model, like what data to generate, how to process it, and which model architecture to apply, directly determine whether the resulting model generalizes to novel biology or simply memorizes the training set. The exact process depends on the specific data modalities included (e.g., transcriptomics, Cell Painting, or multiomics combinations) and the desired outcomes.
The core workflow for training on transcriptomic data is to generate perturbation datasets at scale, clean and preprocess data usually for differential expression between conditions to generate gene signatures of significantly responsive genes, then apply the desired machine learning model, followed by validation on an independent dataset (8-10). Regardless of the data modality used for training, it is becoming clear that AI model efficacy largely depends on the scale, standardization, and perturbation coverage of the training dataset.
How Many Transcriptomic Samples are Required to Train AI Models?
There is no single, most appropriate number of transcriptomic samples for AI model training, because the requirement depends on multiple factors:
- The type of model (supervised vs. generative)
- The predictive task (MoA prediction, toxicity classification, perturbation-response prediction)
- The biological diversity captured (cell types, doses, time points)
- The noise and standardization of the dataset
Traditional methods for generating bulk RNA-seq data are too expensive and time-consuming to produce large-scale, homogeneous perturbation datasets, so few studies have directly evaluated the optimal sample size required to train AI models on this type of data (9).
Given the sparsity of this evidence, it’s valuable to extrapolate from the field of single-cell RNA-seq (scRNA-seq) AI model training despite their different data structures, noise profiles, and information content. Vast, relatively uniform scRNA-seq datasets are available and some of the most powerful AI inference models, such as scGPT, were trained on over 33 million cells and are suited to cell type annotation, multi-batch integration, multi-omic integration, perturbation response prediction, and gene network inference with some modifications (11).
Together, these findings suggest that millions of bulk transcriptome profiles are likely required to train robust, multipurpose predictive models, which can only be generated using ultra-high-throughput methods such as MERCURIUS™ DRUG-seq.
Here’s how MERCURIUS™ DRUG-seq uses massive sample multiplexing for industrial-scale transcriptomics in drug discovery.
The Types of Data Used to Train AI Models in Drug Discovery
While the optimal type of training data depends on the biological question and end-stage predictive model use, omics modalities, such as transcriptomics, genomics, and proteomics, usually form the backbone of many AI models, either individually or together in a multi-omic approach (10). Integration of omics with the high-content screening method, Cell Painting, is also gaining traction, as the combination provides multi-level systems-based insights that are impossible to achieve with each modality alone (12).
The use of transcriptomics and Cell Painting is due to their rich, large-scale, multidimensional readouts, which provide millions of internal data points crucial for inferring disease-related molecular patterns, causal relationships, and toxicogenomic signatures.
Unlike genomics, which reflects relatively static sequence-level information, and proteomics, which captures slower translational changes, the transcriptome responds rapidly in response to perturbations. This makes gene expression data exceptionally well-suited for training AI models that need to learn how biological systems respond to drug-like compounds for detecting early toxicity and derisking pipelines (7).
Learn more about how combining high-throughput transcriptomics with Cell Painting can advance drug discovery and development.
Why is Transcriptomics Ideal for Predictive Model Training in Pharma and What Data is Available?
Transcriptomics is arguably one of the most informative omics layers for drug discovery and AI model training. Because it captures early causal responses to perturbations, it provides the mechanistic depth needed for AI to infer mechanisms of action, target engagement, off-target effects, and emerging toxicity before phenotypic changes become visible (13).
However, while large-scale publicly available bulk RNA-seq datasets, such as the GTEx Consortium, the Cancer Genome Atlas (TCGA), the ENCODE Project, and perturbation signatures from the LINCS Consortium, have advanced traditional and AI-driven discovery, most were developed without AI model training in mind (Table 1). As a result, many datasets are constrained by inconsistent metadata, a lack of dose–response information, heterogeneous protocols, and the absence of standardized perturbation libraries, all of which limit their utility for training predictive models (14).
Similarly, smaller, more bespoke perturbation training datasets are often highly dimensional but have small sample sizes relative to the number of features. This leads to overfitting and poor generalization in AI models without very large datasets (9).
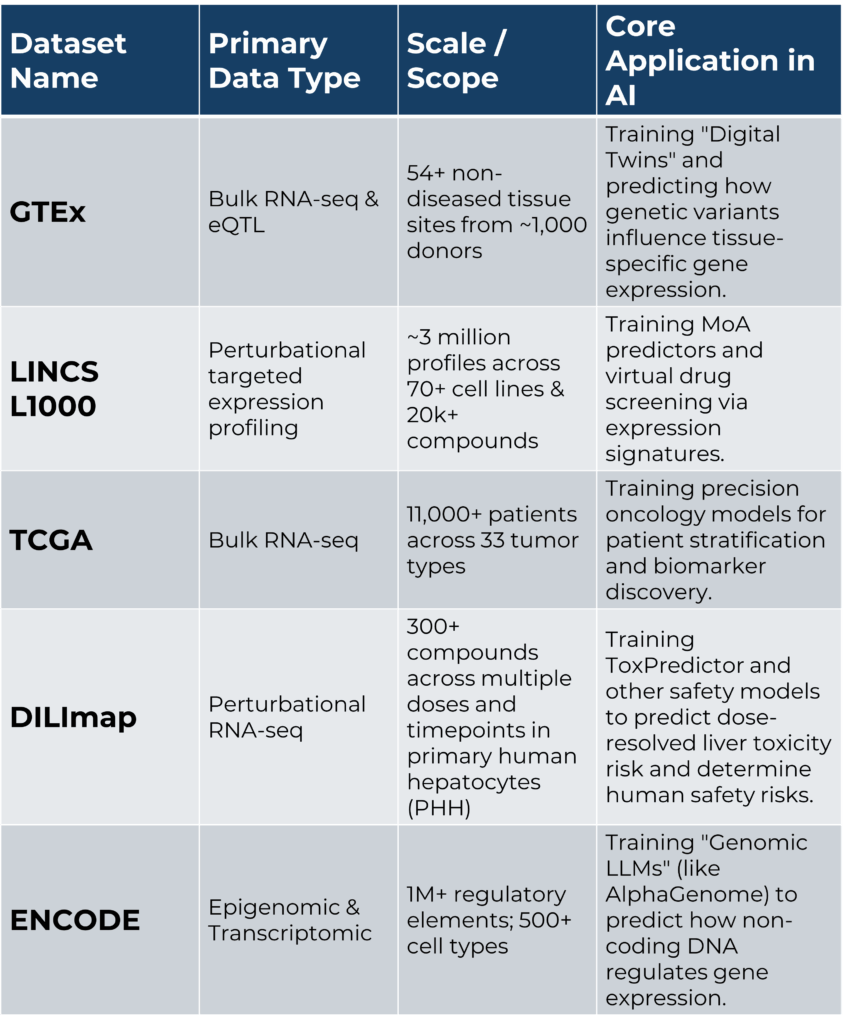
Table 1: Overview of large-scale transcriptomic datasets used to train AI models.
The Main Challenges with Current Transcriptomic Datasets for Training AI Models
Hidden Quality Imbalances, Poor Reproducibility, and Incomplete Metadata
A study of 40 publicly available disease-related RNA-seq datasets found sample quality imbalances in ~35% of datasets, which directly affected differential expression and reproducibility (14). This systematic data-quality bias can confound AI models trained on publicly available bulk RNA-seq datasets. Most also have poor metadata that omits core information, such as dosage, time, and other culture-, technical-, or treatment-related information (14).
Batch Effects and Cross-Study Heterogeneity
Pervasive batch effects arise from variations in sample preparation, library construction, sequencing platforms, and other technical factors. While data preprocessing can attempt to reduce systemic variation and harmonize datasets, underlying heterogeneity can severely limit cross-dataset predictive performance. This is a core problem for AI models intended to generalize (15).
Most Public Data Comes from Non-Perturbed or Healthy/Disease Baseline Samples
Most large-scale RNA-seq resources, such as GTEx, TCGA, and ENCODE, focus on healthy tissue, bulk tumor resections, or non-perturbed immortalized cell lines. AI models trained on baseline-only datasets cannot learn how gene networks change under stress, small-molecule or genetic perturbation, target inhibition, or other dynamic contexts.
When perturbation datasets are available, they rarely cover the whole transcriptome. For example, LINCS L1000 uses targeted expression profiling of only 978 genes and relies on computational inference to estimate expression for the remaining 20,000 genes, thereby missing ground-truth data and novel mRNA targets or biomarkers (16).
Lack of Relevant Sample Types, Perturbations, and Limited Physiologically Appropriate Models
AI model training relies on transcriptomic data from physiologically relevant 2D primary cells or 3D spheroids, which aren’t included in most data repositories. Therefore, the datasets provide limited insight into relevant mechanisms of drug response, pathway dynamics, or causal biology, all of which are needed by AI models to improve predictive accuracy.
For instance, while LINCS L1000 contains approximately 1.3 million perturbation profiles across more than 70 human cell lines, a small number of cell lines like MCF7 and PC3 dominate the dataset, resulting in a highly imbalanced data distribution (13). Similarly, only around 12% of cell lines have sufficient coverage for large-scale pharmacogenomic analysis (13).
Population and Demographic Bias
One study reports a 4.1-fold overrepresentation of donors of European ancestry relative to the global population in bulk RNA-seq datasets on the Sequence Read Archive (SRA). Therefore, for AI models trained on publicly available transcriptomic data, there’s a risk of systemic bias that fails to recognize disease mechanisms or compound safety concerns in underrepresented subpopulations or may not generalize across real patient populations (17).
The Main Challenges with Traditional Sample-by-Sample Bulk RNA-seq Methods for Generating Training Data for AI
While traditional RNA-seq is suited to small-scale studies, numerous challenges arise when pharma screening teams try to increase sample sizes beyond tens or hundreds of samples:
Too Slow, Low-Throughput, and Expensive to Scale for AI-Ready Datasets
Traditional RNA-seq workflows require RNA extraction, individual-sample library preparation, and per-sample handling in separate tubes. These stages, especially RNA extraction, are far too slow for industrial-scale screening and for generating AI training data of sufficient size (18). Similarly, these excessive processing steps make scaling to hundreds of thousands or millions of samples prohibitively costly. Traditional RNA-seq ultimately creates datasets that are far too small to train generalizable models.
High Technical Variability and Batch Effects
Owing to the per-sample processing, variation and sample loss can occur at every step. This variability can introduce significant batch effects in screens of hundreds of thousands of samples, such that even with normalization and batch correction, AI models trained on these data may learn technical noise rather than biology, thus harming generalization (15).
Difficult to End-to-End Automate
While it is possible to automate RNA extraction and sequencing library preparation with liquid-handling instruments, the number of steps means the process remains expensive, laborious, and relatively low-throughput.
For instance, one study of only 84 samples reported that, despite automation, it still took 9 hours to prepare RNA-seq libraries (18). This didn’t include the time-consuming manual RNA extraction steps, thus highlighting the challenge of automating standard RNA-seq approaches to generate datasets of the size required to effectively train AI models.
Want to see what AI-Ready High-Throughput Transcriptomic Data Actually Looks Like? Download our free DRUG-seq benchmark datasets from multiple cell lines.
How MERCURIUS™ DRUG-seq Helps Pharma Generate Transcriptomic AI Training Data on an Industrial Scale
These challenges with publicly available AI training resources and traditional sample-by-sample RNA-seq methods mean that pharma now aims to generate bespoke, proprietary large-scale datasets aligned with the features and goals of its discovery pipeline to produce more refined or generalizable AI model outputs.
Most publicly available transcriptomic training data were generated by multicenter consortia and required years, if not decades, to fully realize, at prohibitive cost given the large number of samples included.
Now, the ultra-high-throughput bulk 3’ mRNA transcriptomics technology, MERCURIUS™ DRUG-seq, helps pharma companies to generate bespoke transcriptomic AI training datasets from tens of thousands of 2D cellular samples within the same screening experiment in a fraction of the time and cost of other methods, without RNA extraction (Fig. 1).
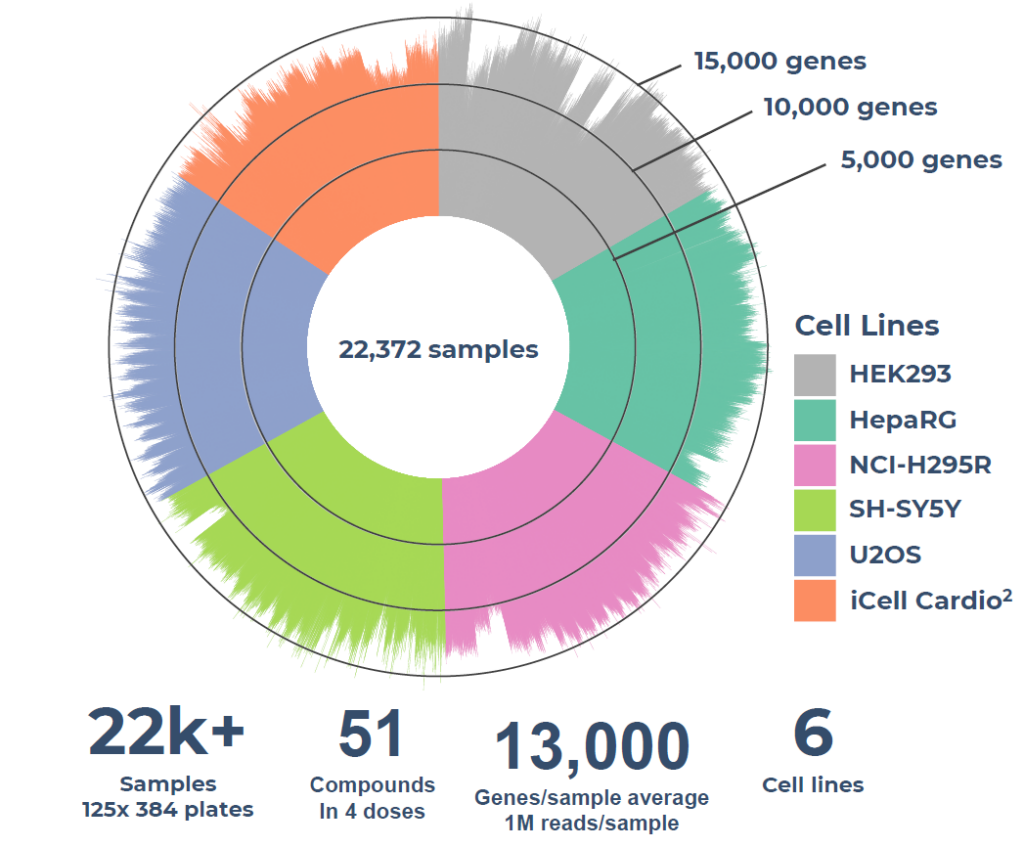
Figure 1. MERCURIUS™ DRUG-seq gene detection overview of a case study on over 22,000 samples and 51 compounds in four doses, on 6 cell lines, sequenced at 1M reads per sample, demonstrating the throughput and consistency required for AI-ready perturbation dataset generation.
Contrary to individual sample library preparation, MERCURIUS™ DRUG-seq’s early sample barcoding and pooling strategy allows massive multiplexing of samples early in the workflow and was designed for automation.
Our kits are available in 96- and 384-well plate formats and are fully automatable using existing screening infrastructure in 1536-well plate format (currently in development). While MERCURIUS™ DRUG-seq provides 3’ readouts of gene expression highly correlated to standard RNA-seq approaches, for those interested in training AI models with splicing, non-coding transcripts, fusion genes, or novel isoforms, we recommend MERCURIUS™ Total DRUG-seq instead.
As 2D cells often lack physiological relevance and as the use of 3D spheroid and organoid models increases in the pharmaceutical discovery and development pipeline, we developed MERCURIUS™ Spheroid DRUG-seq to enable screening-scale transcriptomics for samples that provide more representative cellular environments, interactions, and signaling.
Conclusion: Bespoke, Proprietary Large-Scale Transcriptomic Datasets Provide a Competitive AI-Training Advantage
Public datasets aren’t built for AI training and are too heterogeneous, sparse in perturbations, and biased in their sample composition to robustly train models that reliably generalize across the complexity of discovery pipelines. Similarly, traditional RNA-seq methods are incapable of generating the hundreds of thousands to millions of standardized perturbation expression profiles that robust AI training demands, at a feasible cost or timescale.
High-throughput transcriptomic technologies like MERCURIUS™ DRUG-seq stand to benefit pharma by helping them move from data scarcity and public dependence to proprietary, scalable, and AI-ready data generation.
By enabling thousands of perturbations to be profiled in a single standardized experiment, MERCURIUS™ DRUG-seq provides the throughput, reproducibility, and cost-efficiency required to close the data-generation gap. Those who generate large-scale, high-quality proprietary transcriptomic datasets will almost certainly hold a compounding competitive advantage in future predictive discovery.
If you’re evaluating whether high-throughput transcriptomics is appropriate for your AI training dataset, our team can help you assess fit and feasibility tailored to your cell type, compound library, and model objectives.
References
- DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: new estimates of R&D costs. Journal of Health Economics. 2016 May 1;47:20-33.
- Mullard A. The drug-maker’s guide to the galaxy. Nature. 2017 Sep 28;549(7673):445-7.
- Steyaert S, Pizurica M, Nagaraj D, Khandelwal P, Hernandez-Boussard T, Gentles AJ, Gevaert O. Multimodal data fusion for cancer biomarker discovery with deep learning. Nature Machine Intelligence. 2023 Apr;5(4):351-62.
- Pun FW, Ozerov IV, Zhavoronkov A. AI-powered therapeutic target discovery. Trends in Pharmacological Sciences. 2023 Sep 1;44(9):561-72.
- Mullowney MW, Duncan KR, Elsayed SS, Garg N, van der Hooft JJ, Martin NI, Meijer D, Terlouw BR, Biermann F, Blin K, Durairaj J. Artificial intelligence for natural product drug discovery. Nature Reviews Drug Discovery. 2023 Nov;22(11):895-916.
- Banerjee J, Taroni JN, Allaway RJ, Prasad DV, Guinney J, Greene C. Machine learning in rare disease. Nature Methods. 2023 Jun;20(6):803-14.
- Bergen V, Kodella K, Srikrishnan S, Barrandon O, Anderson S, Rogers-Grazado M, Fowler C, Beyene H, Robichaud N, Fulton T, Lapchyk N. A large-scale human toxicogenomics resource for drug-induced liver injury prediction. Nature Communications. 2025 Nov 13;16(1):9860
- Cheng Y, Xu SM, Santucci K, Lindner G, Janitz M. Machine learning and related approaches in transcriptomics. Biochemical and Biophysical Research Communications. 2024 Sep 10;724:150225.
- Silvey S, Olex A, Tang S, Liu J. Sample size requirements for machine learning classification of binary outcomes in bulk RNA-Seq data. BMC Bioinformatics. 2026 Jan 31.
- Zhang K, Yang X, Wang Y, Yu Y, Huang N, Li G, Li X, Wu JC, Yang S. Artificial intelligence in drug development. Nature Medicine. 2025 Jan;31(1):45-59
- Cui H, Wang C, Maan H, Pang K, Luo F, Duan N, Wang B. scGPT: toward building a foundation model for single-cell multi-omics using generative AI. Nature Methods. 2024 Aug;21(8):1470-80.
- Seal S, Trapotsi MA, Spjuth O, Singh S, Carreras-Puigvert J, Greene N, Bender A, Carpenter AE. Cell Painting: a decade of discovery and innovation in cellular imaging. Nature methods. 2025 Feb;22(2):254-68.
- Chen H, King FJ, Zhou B, Wang Y, Canedy CJ, Hayashi J, Zhong Y, Chang MW, Pache L, Wong JL, Jia Y. Drug target prediction through deep learning functional representation of gene signatures. Nature communications. 2024 Feb 29;15(1):1853.
- Sprang M, Möllmann J, Andrade-Navarro MA, Fontaine JF. Overlooked poor-quality patient samples in sequencing data impair reproducibility of published clinically relevant datasets. Genome Biology. 2024 Aug 16;25(1):222.
- Van R, Alvarez D, Mize T, Gannavarapu S, Chintham Reddy L, Nasoz F, Han MV. A comparison of RNA-Seq data preprocessing pipelines for transcriptomic predictions across independent studies. BMC bioinformatics. 2024 May 8;25(1):181.
- Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK, Lahr DL. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017 Nov 30;171(6):1437-52.
- Romero IG, Rodenberg G, Arner AM, Li L, Beasley IJ, Rossow R, Ryan N, Wang S, Lea AJ. Public RNA-seq data are not representative of global human diversity. bioRxiv. 2024 Oct 12:2024-10.
- Santacruz D, Enane FO, Fundel-Clemens K, Giner M, Wolf G, Onstein S, Klimek C, Smith Z, Wijayawardena B, Viollet C. Automation of high-throughput mRNA-seq library preparation: a robust, hands-free and time efficient methodology. SLAS Discovery. 2022 Mar 1;27(2):140-7.