
Full-Length BRB-seq is a rapid, cost-effective, massively multiplexed bulk mRNA-seq library preparation method that harnesses poly(A) tailing, sample barcoding, and early sample pooling to provide users with robust transcriptome-wide data across the entire length of transcripts for up to 96 samples simultaneously. Our technology meets the demand for comprehensive ultra-high-throughput transcriptomic solutions that accelerate diverse pipelines, including drug discovery and development, toxicogenomics, biomarker discovery, agrogenomics, and basic research.
But how does Full-Length BRB-seq achieve this unparalleled scalability and cost-effectiveness, and how does it compare to standard, non-pooled full-length mRNA-seq methods?
In this blog post, we explain the ins and outs of Full-Length BRB-seq and demonstrate its power to generate sequencing reads across entire transcripts for over 50,000 isoforms comparable to non-multiplexed competitor mRNA-seq.
Born from BRB-seq: 3’ mRNA-seq goes full-length
Full-Length BRB-seq was born from our Bulk RNA Barcoding and sequencing (BRB-seq) method, in which highly optimized barcoded oligo(dT) primers uniquely tag the 3’ poly(A) tail of each individual mRNA per sample. This barcoding strategy allows users to multiplex samples early in the workflow, maximizing throughput while reducing hands-on time and cost.
While BRB-seq provides highly robust transcriptome-wide gene expression information comparable to standard non-multiplexed mRNA-seq methods, it targets the 3’ region of mRNA but not the whole transcript as non-3’ fragments lack a barcode and thus cannot be amplified or sequenced, meaning splice variants and transcript isoforms are missed.
To address this, we developed Full-Length BRB-seq, which combines the scalability and cost-effectiveness of BRB-seq with entire transcript read-outs for researchers requiring splicing and isoform-level information, alongside overall gene expression levels.
An Overview of Full-Length BRB-seq
Full-Length BRB-seq has a one-day lab workflow that leverages our advanced BRB-seq barcoding technology, with some additional steps at the start of the protocol to enable the sequencing of full transcripts and their reconstruction during data analysis.
The main protocol additions include the early fragmentation of full-length mRNA molecules, repair of the RNA fragments, and RNA fragment poly-adenylation. These steps are the foundation of Full-Length BRB-seq because they ensure that each fragment gains a poly-A tail and can therefore be barcoded with our highly optimized oligo(dT) primers containing unique molecular identifiers (UMIs) to distinguish between original mRNA fragments and amplification duplicates, unique sample barcodes that assign distinct identifiers to each individual RNA sample, and adaptors for primer annealing. These barcodes allow for sample demultiplexing after sequencing.
The Full-Length BRB-seq protocol requires around 4 hours and 20 minutes of hands-on time and 10-1000 ng of purified total RNA per sample with RIN greater than six and Nanodrop A260/280 ratios greater than 1.5. RNA quantity and quality must be uniform across all samples to ensure an even distribution of reads after sequencing.
With our 96-sample kit, users can multiplex up to 96 samples in the same tube or can prepare up to 96 samples distributed across up to four different libraries. We recommend a sequencing depth of between 12-20 million reads per sample to enable the reliable and unbiased detection of over 20,000 genes, including those with low expression. However, if only highly expressed genes are of interest, sequencing depths as low as five million reads per sample can still generate comprehensive data but allow more sample multiplexing. Depending on sequencing capacity and individual requirements, deeper sequencing will generally reduce the number of samples it is possible to multiplex per tube. Full-Length BRB-seq is compatible with Illumina® and AVITI™ sequencers.
The Full-Length BRB-seq Workflow
Firstly, mRNA is enriched from each purified total RNA sample by oligo-dT-based purification, followed by fragmentation, which requires 90-120 minutes of hands-on time (Fig. 1 – step 1). Fragmentation of these enriched full-length mRNAs is crucial to ensure that all fragment sequences along the entire transcript can be poly-adenylated in the following step. Poly-adenylating all RNA fragments is necessary for sample barcoding along the whole length of transcripts.
The next step repairs these purified mRNA fragments, poly-adenylates them if they lack poly-A tails, and reverse transcribes the poly-adenylated fragments with barcoded oligo(dT) primers in first-strand synthesis. After this point, barcoded samples are pooled into the same tube and processed simultaneously for all subsequent steps. This stage requires only 15-20 minutes of hands-on time (Fig. 1 – step 2).
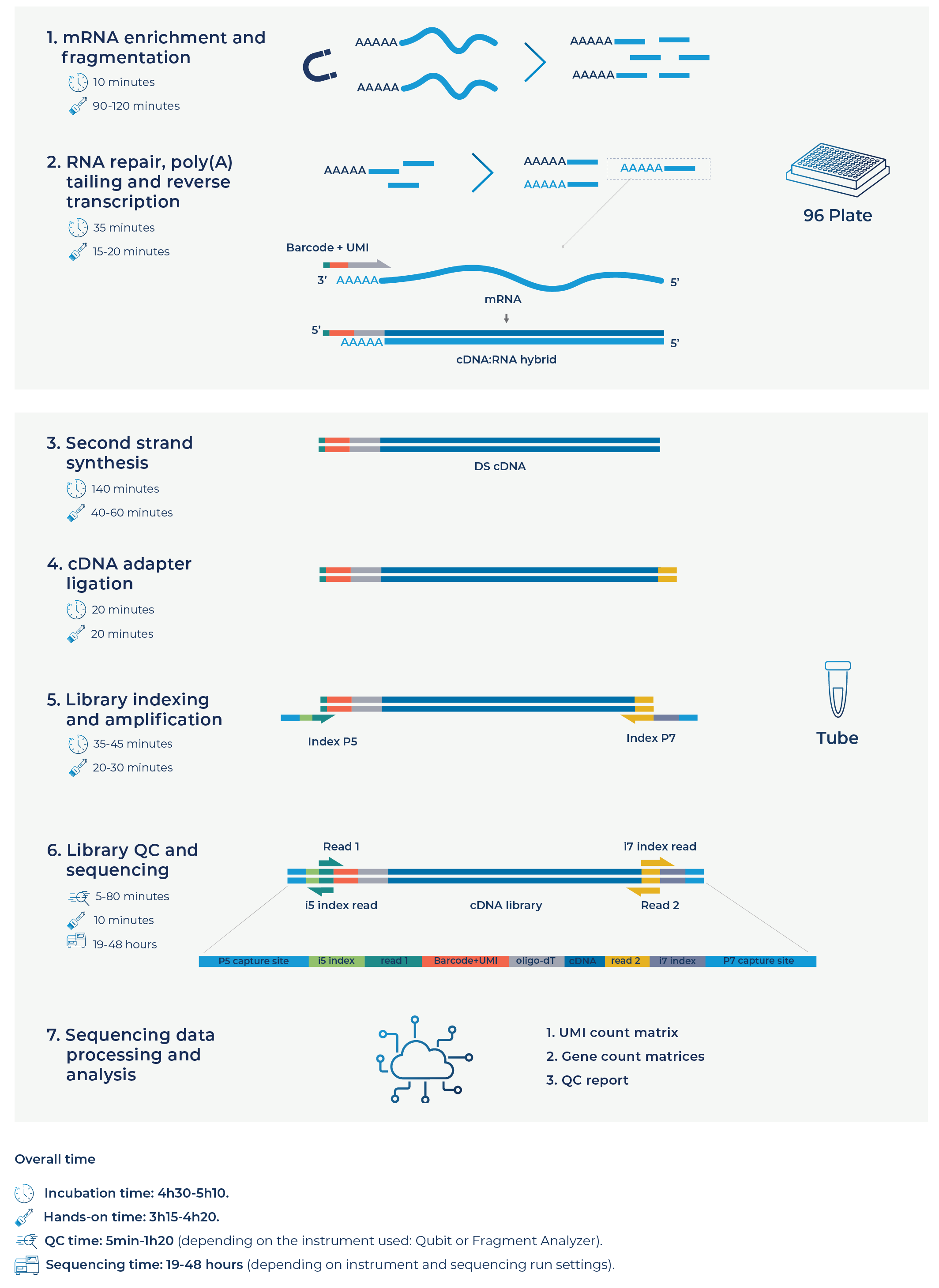
Figure 1. The Full-Length BRB-seq workflow.
From here on, the pooled samples undergo second-strand synthesis and ligation of adapters to the double-stranded cDNA, followed by library indexing, amplification, and quality control, similar to the BRB-seq protocol.
After sequencing at the desired read depth per sample, raw data is demultiplexed using the barcode set located on the kit label and processed to generate UMI count matrices, gene count matrices, and quality control reports for each individual sample. The processed data is then ready for downstream investigation, such as the detection of splice variants and transcript isoforms, or isoform differential expression analysis between different experimental conditions, drug treatments, cell types, or disease samples.
High Demultiplexing and Mapping Rates for Full-Length BRB-seq
We have extensively validated Full-Length BRB-seq to ensure it provides the most accurate, robust, and comprehensive gene expression data possible.
For instance, data generated with Full-Length BRB-seq of 48 pooled samples sequenced at 12 million reads had a read demultiplexing rate of over 99%, an exonic mapping rate of almost 80%, and a read duplication rate of only 18% (Fig. 2).
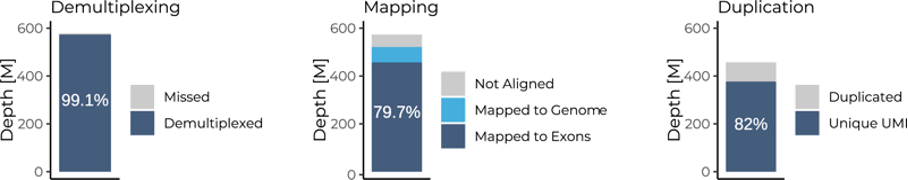
Figure 2. Barplots indicating the demultiplexing, mapping, and duplication rates for 48 samples sequenced at 12 million reads per sample. ([M] – million reads).
This high demultiplexing and mapping rate allowed the uniform detection of over 22,000 genes for all 48 samples, ensuring excellent transcriptome coverage (Fig. 3).

Figure 3. Barplot indicating the number of detected genes for each of the 48 samples prepared and multiplexed with Full-Length BRB-seq and sequenced at 12 million reads per sample. (CPM – Counts per million).
Full-Length BRB-seq is Highly Comparable to Non-Multiplexed Full-length mRNA-seq
We have extensively benchmarked Full-Length BRB-seq against standard BRB-seq and a standard non-multiplexed full-length mRNA-seq method to ensure that reads are robustly generated across the entire length of genes and that it can detect similar numbers of transcripts.
Standard BRB-seq showed increased read distribution at the 3’ end of the gene, as expected, due to its poly(A) selection strategy. In contrast, Full-Length BRB-seq showed a consistent and uniform distribution of reads across the entire gene body and was highly comparable to the non-multiplexed full-length mRNA-seq competitor technology (Fig. 4).
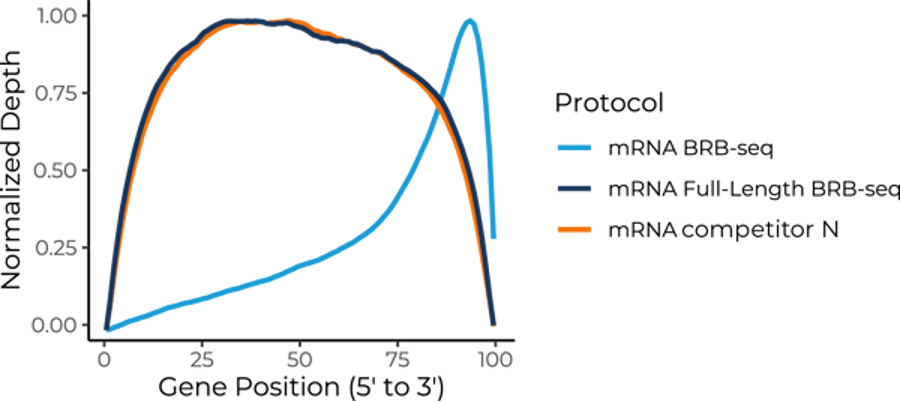
Figure 4. A comparison of the normalized read distribution averaged across all gene bodies shows a uniform and consistent distribution of Full-Length BRB-seq reads across the whole gene, comparable to a non-multiplexed competitor full-length mRNA-seq technology at sequencing depths of between three and four million reads per sample for each approach. Universal Human Reference RNA from ThermoFisher Scientific was used as input for all benchmarking.
This benchmarking experiment also indicated that Full-Length BRB-seq performs similarly to a competitor mRNA-seq technology at all read depths. It was highly effective in detecting over 50,000 transcript isoforms at 12 million reads per sample, with around 48,000 isoforms detected even at a relatively low sequencing depth of five million reads per sample (Fig. 5).
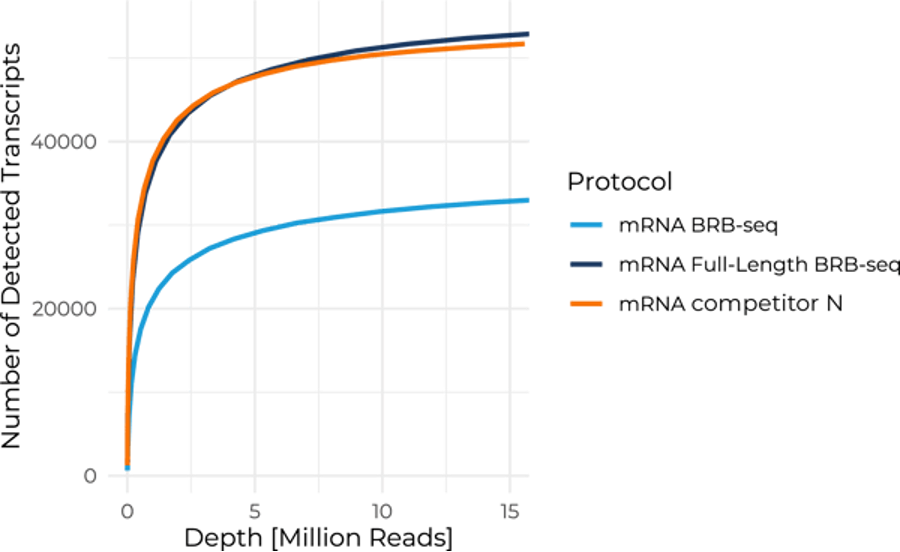
Figure 5. Number of detected transcripts for Full-Length BRB-seq compared to standard BRB-seq and a non-multiplexed full-length mRNA-seq competitor. Full-Length BRB-seq and the full-length mRNA-seq competitor detect comparable numbers of transcripts at all read depths.
A Massively Multiplexed Solution for Ultra-High-Throughput Studies
Overall, Full-Length BRB-seq is a massively multiplexed bulk mRNA-seq solution for researchers requiring transcriptional information across the whole length of transcripts to identify splice variants and different isoforms. The one-day workflow is rapid, cost-effective, and scalable to meet ultra-high-throughput needs in drug discovery, toxicogenomics, and beyond.
Contact us to find out more about how our Full-Length BRB-seq service and kit could help with your next discovery.