
Since their development around 30 years ago, TaqMan™ gene expression assays have been a popular choice for scientists requiring highly sensitive and specific readouts of gene activity.
However, despite their utility, TaqMan gene expression assays limit researchers to assessing ~100 bp-long amplicons of relatively small panels of predefined genes. This could present significant bottlenecks in high-throughput screening studies, especially if the particular pathways or genes affected by chemical or genetic perturbations aren’t yet known, or transcriptome-wide information is required for broad exploratory analyses.
So, 30 years on, are your TaqMan gene expression assay screens due for an omics-based upgrade in the era of big data?
TaqMan Assays in a Nutshell
TaqMan gene expression assays rely on a pair of unlabeled primers that generate an amplicon of between 50 and 150 bp, preferably spanning exons to limit genomic DNA amplification. A TaqMan probe labeled with a fluorophore at the 5′ end and a nonfluorescent quencher (NFQ) and minor groove binder (MGB) at the 3′ end binds the region between the primers. The fluorophore is quenched when in proximity to the NFQ.
Upon PCR, the 5´–3´ exonuclease activity of Taq polymerase degrades the probe, releasing the fluorophore from the quencher, allowing it to fluoresce upon excitation. Each PCR cycle releases more fluorophore molecules, resulting in an increase in fluorescence intensity that is directly proportional to the amount of amplicon synthesized, as detected by standard real-time qPCR cyclers.
Users can select from predesigned or custom TaqMan assays to detect individual genes in a single tube, microfluidic array cards for 384 simultaneous real-time PCR (qPCR) reactions on up to eight samples, or array plates for 96 samples or fewer than 48 targets.
The TaqMan Gene Expression Assay in Action
Over the decades, researchers have utilized TaqMan technology to compare the expression levels of genes across different conditions, tissues, or cell types, to independently validate RNA-seq or microarray results, to confirm gene silencing or overexpression, and to monitor expression changes following drug treatment, among other applications.
For instance, a team led by researchers from Kanazawa University, Kanazawa, Japan, recently used the TaqMan Array Human Molecular Mechanisms of Cancer, which contains 92 gene assays associated with cancer mechanisms and pathways, to screen for gene expression biomarkers of sensitivity to an epigenome-targeting drug, TAK-981 (subasumstat) [1].
TAK-981 is a promising first-in-class compound that inhibits SUMO-activating enzyme E, which is crucial for a process called SUMOylation and has shown promise in cancer therapeutics [2]. SUMOylation covalently attaches small ubiquitin-like modifier (SUMO) proteins to other proteins and is involved in numerous cellular processes, ranging from transcriptional regulation to apoptosis [3]. The SUMOylation pathway is a promising therapeutic target as it is also implicated in various diseases, including cancer [3].
A Lucky Find in a Predefined TaqMan Cancer Gene Panel
To assess the effect of inhibiting SUMOylation with TAK-981 in cancer cells, the researchers treated the KRAS–mutant human colon cancer HCT116 cell line with 1 µM TAK-981 for 48 hours (Fig. 1A). They then isolated RNA and performed cDNA synthesis, followed by the TaqMan assay, which identified eight upregulated genes and seven downregulated genes (Fig. 1B & C).
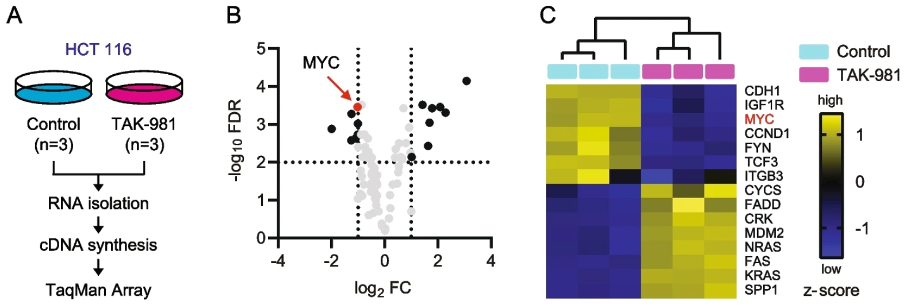
Figure 1. (A) Overview of the TaqMan Array experiment. HCT116 cells were treated with dimethyl sulfoxide as a control or 1 µM TAK-981 for 48 hours (n = 3). (B) volcano plot and (C) normalized heatmap indicating differentially expressed genes FC > I2I and FDR < 0.01. MYC is indicated in red. Figure taken from Kotani et al., 2024 [1].
The oncogene MYC was the most promising sensitivity biomarker as it was significantly downregulated, is a critical mediator of KRAS function, and SUMOylation inhibition is known to regulate MYC-driven tumorigenesis [4].
The TaqMan gene expression screen was supported by an extensive series of functional, biochemical, cellular viability, and high-content imaging assays, which confirmed that TAK-981-mediated downregulation of MYC effectively suppressed the growth of MYC-expressing KRAS-mutant cancers across different tissue types. When used in combination with another drug called trametinib, TAK-981 caused cell death in multiple cancer cell lines, mouse-derived tumor organoids, and long-term tumor regression in mouse xenograft models.
Is 0.46% of the Transcriptome Enough?
While these results are undoubtedly robust, the TaqMan gene expression screen represented a mere 0.46% of the coding genome (92/20,000 genes). Fortunately, in this instance, the researchers found that a core regulator of tumorigenesis, MYC, was affected as it was included in the TaqMan panel.
However, the screen provided extremely limited insights into other transcriptomic effects of TAK-981 treatment. As SUMOylation has widespread functional roles in many cellular processes, it is plausible that TAK-981 has other direct or indirect transcriptomic effects in non-cancer pathways that may contribute to the observed cellular phenotypes but that were missed with the small panel of genes investigated.
A more thorough “whole-picture” approach would be to use bulk RNA-seq methods, such as MERCURIUS™ DRUG-seq or MERCURIUS™ BRB-seq, which provide unbiased transcriptome-wide readouts not limited to small panels of pathway- or disease-specific genes. Broader overviews would provide fundamental insights impossible to achieve with targeted methods like TaqMan gene expression assays or other probe-based assays like TempO-seq™.
A Broader Dynamic Range and More Samples with Omics
RNA-seq-based approaches would also have helped the authors overcome one of the main limitations they stated about their study:
“Our limitations are that we used a biased approach to the analysis and were unable to define a cut-off value for MYC expression.”
Approaches like MERCURIUS™ DRUG-seq and MERCURIUS™ BRB-seq are unbiased and benefit from a broader dynamic range than PCR-based methods, making it easier to obtain more robust cut-offs for biomarker expression in response to drug treatment. Unlike RT-qPCR, omics approaches also don’t require data normalization to one or a handful of ‘housekeeping’ genes, the choice of which can skew results depending on the gene selected.
TaqMan assays also constrain the user to assessing relatively low numbers of samples simultaneously compared to the 384-sample capacity of MERCURIUS™ technologies for truly scalable screens. A higher number of replicates would have made the findings even more robust.
Transcriptomics Provides a Cost-Effective, Whole-Picture View at Scale
Overall, while TaqMan gene expression arrays are suitable for hypothesis testing or validation, they are not optimal for broader exploratory screening with multiple compounds, experimental conditions, or “whole-picture” studies.
Furthermore, scalable omics technologies now provide transcriptome-wide gene expression information at a similar cost to profiling a handful of genes using reverse transcription quantitative PCR (RT-qPCR), making them cost-effective options for budget-conscious screening without compromising on sample and data throughput [5].
Contact us to discover more about how your compound screen and drug discovery pipeline can benefit from an omics upgrade.
Reference
- Kotani H, Oshima H, Boucher JC, Yamano T, Sakaguchi H, Sato S, Fukuda K, Nishiyama A, Yamashita K, Ohtsubo K, Takeuchi S. Dual inhibition of SUMOylation and MEK conquers MYC-expressing KRAS-mutant cancers by accumulating DNA damage. Journal of Biomedical Science. 2024 Jul 11;31(1):68.
- Lightcap ES, Yu P, Grossman S, Song K, Khattar M, Xega K, He X, Gavin JM, Imaichi H, Garnsey JJ, Koenig E. A small-molecule SUMOylation inhibitor activates antitumor immune responses and potentiates immune therapies in preclinical models. Science translational medicine. 2021 Sep 15;13(611):eaba7791.
- Hendriks IA, Vertegaal AC. A comprehensive compilation of SUMO proteomics. Nature reviews Molecular cell biology. 2016 Sep;17(9):581-95.
- Kessler JD, Kahle KT, Sun T, Meerbrey KL, Schlabach MR, Schmitt EM, Skinner SO, Xu Q, Li MZ, Hartman ZC, Rao M. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science. 2012 Jan 20;335(6066):348-53.
- Alpern D, Gardeux V, Russeil J, Mangeat B, Meireles-Filho AC, Breysse R, Hacker D, Deplancke B. BRB-seq: ultra-affordable high-throughput transcriptomics enabled by bulk RNA barcoding and sequencing. Genome biology. 2019 Dec;20:1-5.