
The Smart-seq2 protocol was the gold standard for full-length plate-based single-cell RNA-seq, offering superior sensitivity and the transcript coverage necessary to detect splice isoforms, allelic variants, and single-nucleotide polymorphisms (SNPs) compared to droplet-based methods (1-3).
However, driven by the success of Smart-seq2, the field continues to evolve rapidly, and newer methods, such as Smart-seq3 and FLASH-seq, provide enhanced performance, speed, cost, and convenience, thanks to protocol upgrades and improved chemistry (4).
So, if you’re a scientist currently using full-length plate-based single-cell RNA-seq methods, in this article, we provide a comprehensive comparison of the Smart-seq2, Smart-seq3, and FLASH-seq protocols to help you evaluate the advantages of transitioning to FLASH-seq, particularly given its availability as both kit and service options from Alithea Genomics.
Smart-seq2 and Smart-seq3 vs. FLASH-seq at a Glance
Before diving into the protocol details, here’s a quick overview of how Smart-seq2, Smart-seq3, FLASH-seq, and MERCURIUS™ FLASH-seq compare across key performance and usability metrics (Table 1).
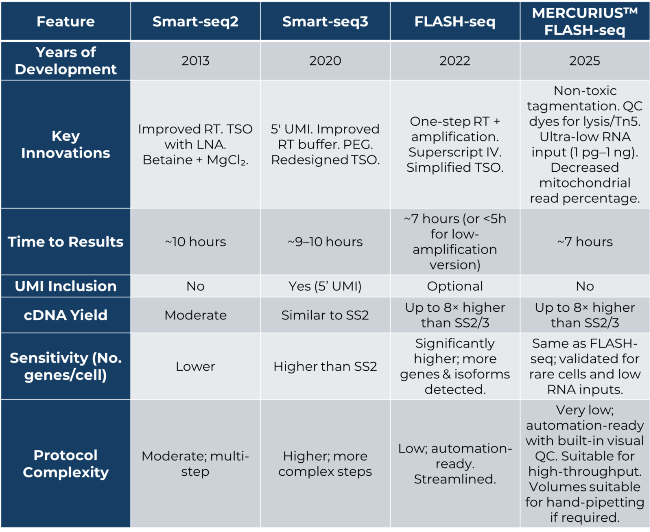
Table 1. Comparison of key features and metrics of each method.
Smart-seq2: The old full-length single-cell standard
Published in 2013, the Smart-seq2 protocol was initially developed by Simone Picelli and colleagues in the lab of Rickard Sandberg at the Ludwig Institute for Cancer Research, Stockholm, Sweden (1). It directly addressed the coverage, sensitivity, and throughput limitations faced by other full-length single-cell RNA-seq methods by building upon the team’s original Smart-seq protocol from 2012 (5). Smart-seq2 benefited from improved reverse transcription, template switching, and preamplification steps to increase the length and yield of cDNA libraries generated from individual cells (1).
The protocol optimized several key parameters, including the use of locked nucleic acid (LNA) guanylate at the 3’ end of the template-switching oligonucleotide (TSO) and the addition of the methyl group donor betaine with higher MgCl₂ concentrations (1). These modifications resulted in a twofold increase in cDNA yield compared to a commercially available Smart-seq protocol, enabling researchers to detect more genes that were potentially crucial to their study (1).
Smart-seq3: Adding Sensitivity with UMI Integration
In 2020, the Sandberg lab developed Smart-seq3, introducing multiple improvements to the Smart-seq2 protocol (3). Arguably, the most significant of these improvements was the inclusion of 5’ unique molecular identifiers (UMI), to allow users to control for PCR amplification biases in silico while maintaining full-length transcript coverage.
The team also completely revised the RT mix as they utilized Maxima H-minus reverse transcriptase for enhanced sensitivity, replaced KCl with NaCl during reverse transcription to reduce RNA secondary structures, added 5% polyethylene glycol to increase beneficial molecular crowding, and also redesigned the TSO with an 11-bp tag sequence and an 8-bp UMI and three riboguanosines to hybridize to the non-templated overhang at the end of single-stranded cDNA (3).
These modifications significantly improved performance, enabling the detection of thousands more transcripts per cell compared to Smart-seq2 and significantly boosted cell-to-cell gene expression profile correlations (3).
FLASH-seq: The Most Sensitive Single-Cell RNA-seq Method Yet
In 2022, Vincent Hahaut and colleagues from the lab of Simone Picelli (the creator of Smart-seq2) at the Institute of Molecular and Clinical Ophthalmology in Basel, Switzerland, published FLASH-seq (4). The team designed the protocol to address the primary limitations of the existing Smart-seq technologies, such as two-day processing times, complex workflows, and suboptimal sensitivity.
FLASH-seq integrates reverse transcription and cDNA amplification into a single step, resulting in a one-day workflow (seven hours) compared to the two-day workflow of Smart-seq2 and Smart-seq3 (nine to ten hours) (Fig. 1). The low-amplification version of FLASH-seq reduces the workflow to less than five hours.
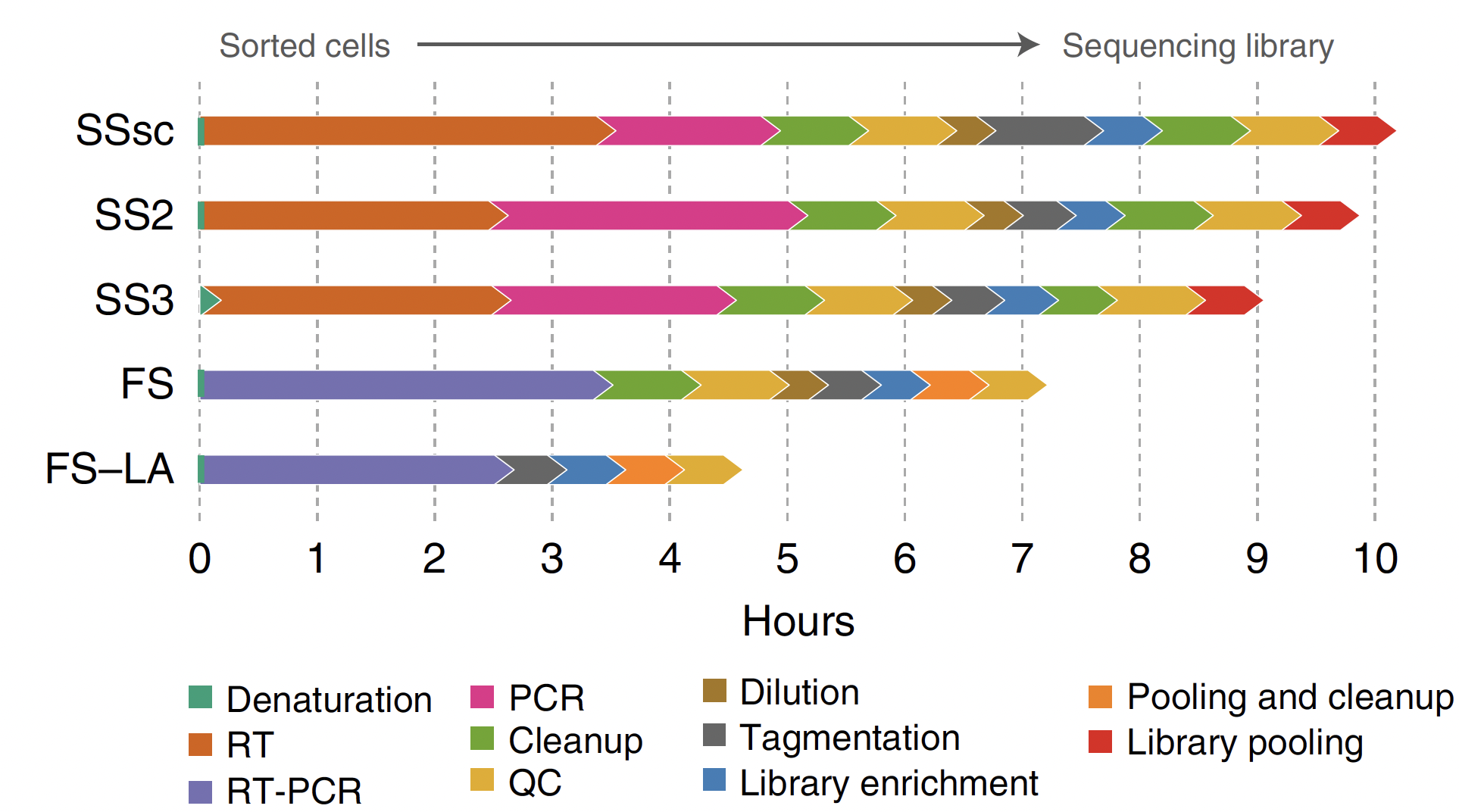
Figure 1. FLASH-seq is up to three hours quicker than Smart-seq protocols. Workflow comparison and estimated time to process a 96-well plate of HEK293T cells for SSsc (SMART-Seq Single Cell Kit from Takara), Smart-seq2 (SS2), Smart-seq3 (SS3), FLASH-seq (FS), and FLASH-seq low amplification (FS-LA). Steps are color-coded. QCs include measurements of concentration and size distribution. Figure taken from (4).
FLASH-seq also uses a more processive reverse transcriptase, which provides significantly improved average gene lengths, indicating better full-length coverage of longer transcripts (Fig. 2A). These modifications also led to FLASH-seq yielding eight times more cDNA than the Smart-seq protocols for the same number of PCR cycles; improvements that are crucial for extremely low-input samples like single cells (Fig. 2B).
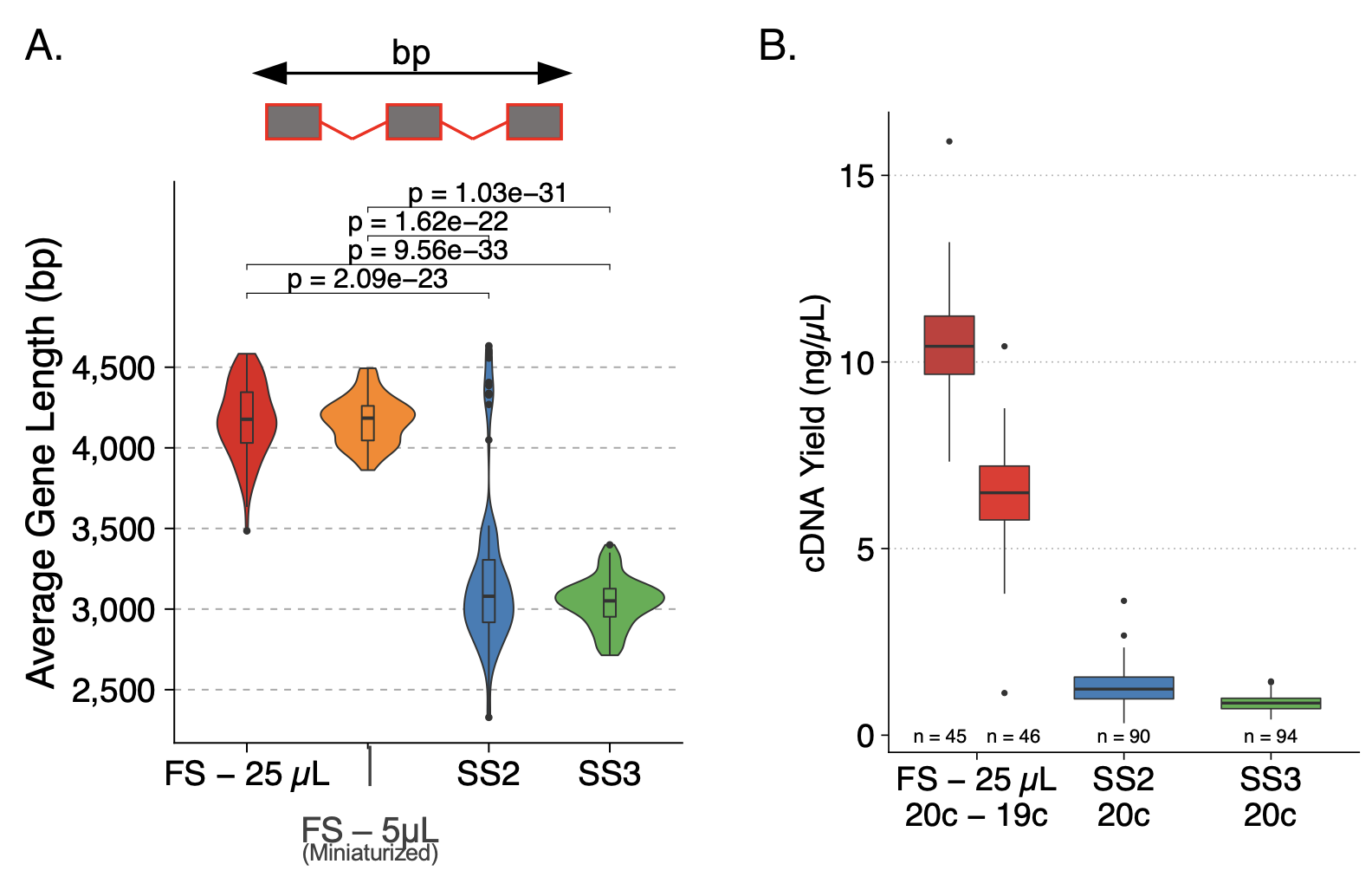
Figure 2. FLASH-seq provides significantly greater full-length coverage for longer genes and generates more cDNA than Smart-seq methods. (A) Plot indicating the average gene length for each method tested. (B) Boxplots indicating the cDNA yield for FLASH-seq at 25 µl and 5 µl volumes compared to Smart-seq2 and Smart-seq3. Statistical testing was performed using a two-sided Dunn’s test, with Bonferroni correction, and adjusted P-values are shown. Figure modified from (4).
Hahaut also increased the dCTP concentration to favor C-tailing activity and boost template-switching efficiency, and simplified the TSO design by replacing LNA guanosine with riboguanosine to reduce strand-invasion artifacts. The published method can be performed with or without the addition of UMIs.
Together, these changes resulted in a significantly higher number of genes and transcripts detected per cell compared to Smart-seq2 and Smart-seq3 (Fig. 3). 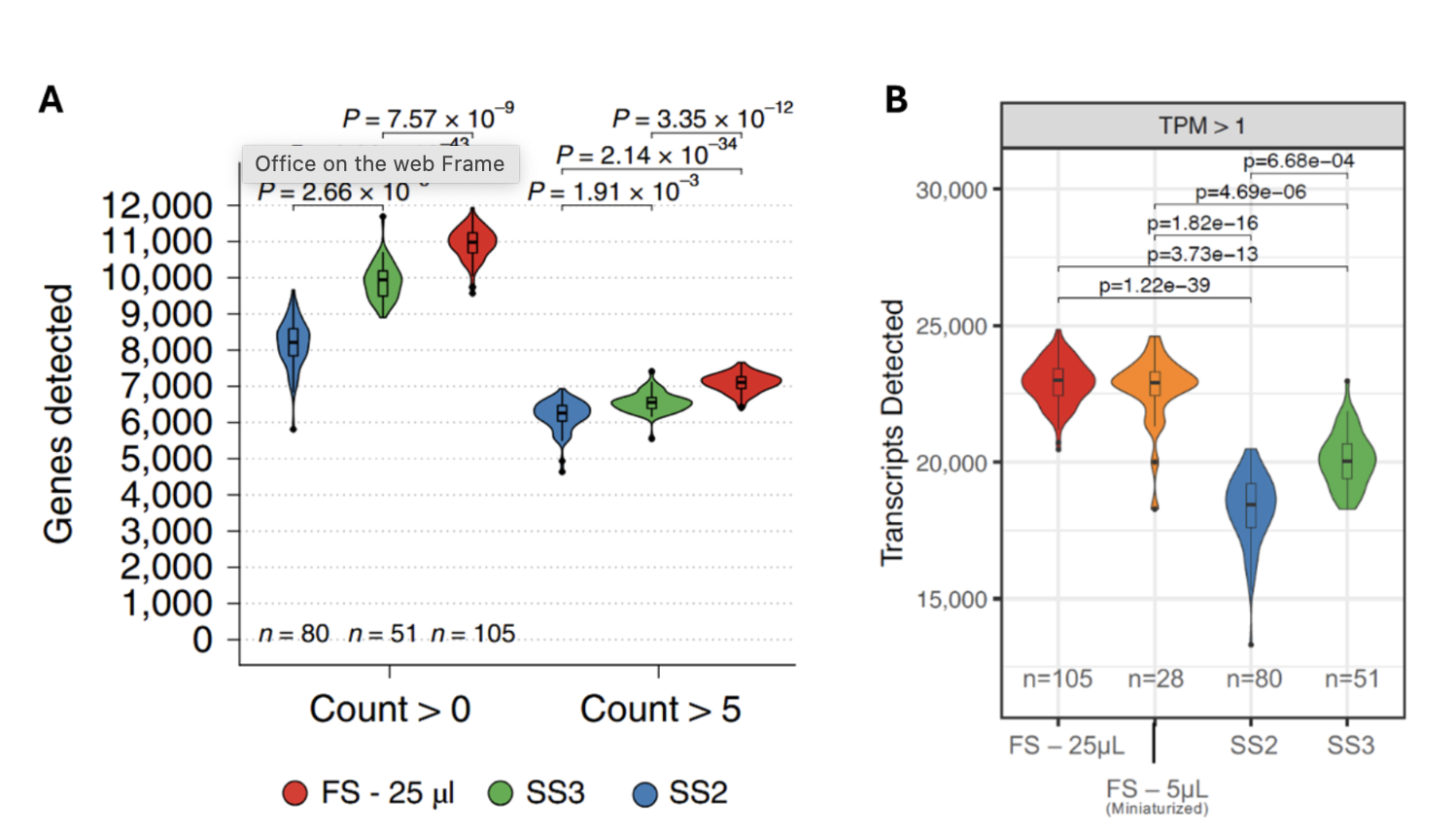
Figure 3. FLASH-seq detects significantly more genes and transcript isoforms than Smart-seq methods. (A) Number of genes detected in HEK293T cells processed by each method at two read thresholds, with reads downsampled to 500,000 raw reads. (B) Number of isoforms detected in the same cells as (A), downsampled to 500K raw reads, with a library-scaled transcript-per-million count (TPM) >1. A two-sided Dunn’s test was used, and Bonferroni-corrected, adjusted P values are shown. Figure modified from (4).
Crucially, FLASH-seq also showed significantly improved cell-to-cell correlations compared to both Smart-seq methods, indicating high technical reproducibility, low variability, and low background noise, each of which is essential for more accurate identification of cell types and detection of subtle biological differences that might be missed otherwise (Fig. 4).
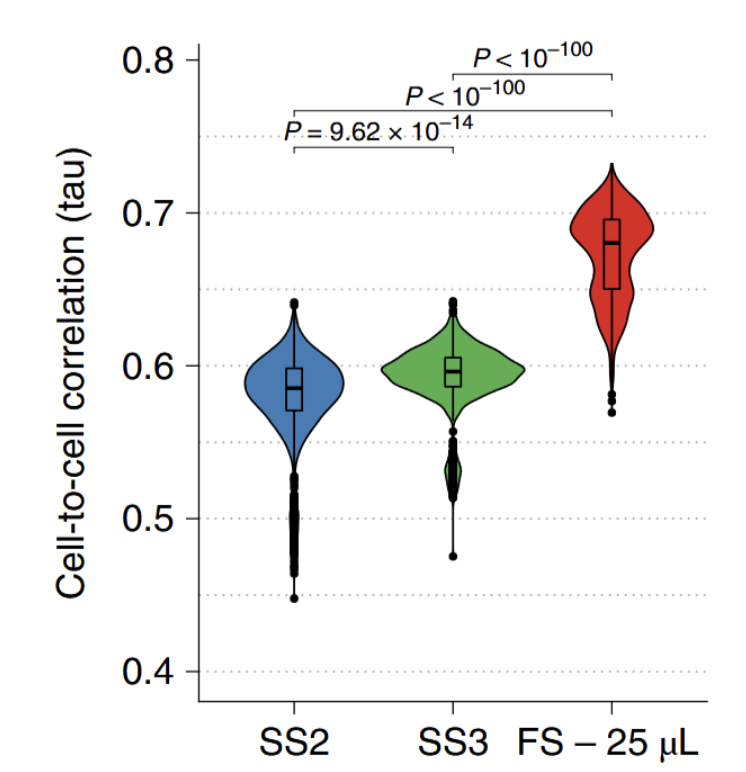
Figure 4. FLASH-seq boosts reproducibility compared to Smart-seq methods. Plot indicating cell-to-cell Kendall’s tau correlations among gene expression in each method using only genes expressed in all three methods (number of genes = 20,042). A two-sided Dunn’s test was used, and Bonferroni-corrected, adjusted P values are shown. Figure from (4).
FLASH-seq’s higher sensitivity and reproducibility also favored the capture of a more diverse set of isoforms and genes, particularly protein-coding genes, compared to Smart-seq2 and Smart-seq3, and facilitated the robust sequencing of cells with low RNA content, such as human peripheral blood mononuclear cells (hPBMCs), alongside the robust detection of SNPs.
FLASH-seq data also allows immunology researchers to infer T-cell receptor (TCR) and B-cell receptor (BCR) sequences, including the critical antigen-binding CDR3 amino acid (CDR3aa) region, even at low sequencing depths (Fig. 5). This multimodal information enables users to study clonal diversity and expansion and pair immune receptor identity with the full transcriptomic state of the same cell without requiring high sequencing depth or multiple specialized protocols.
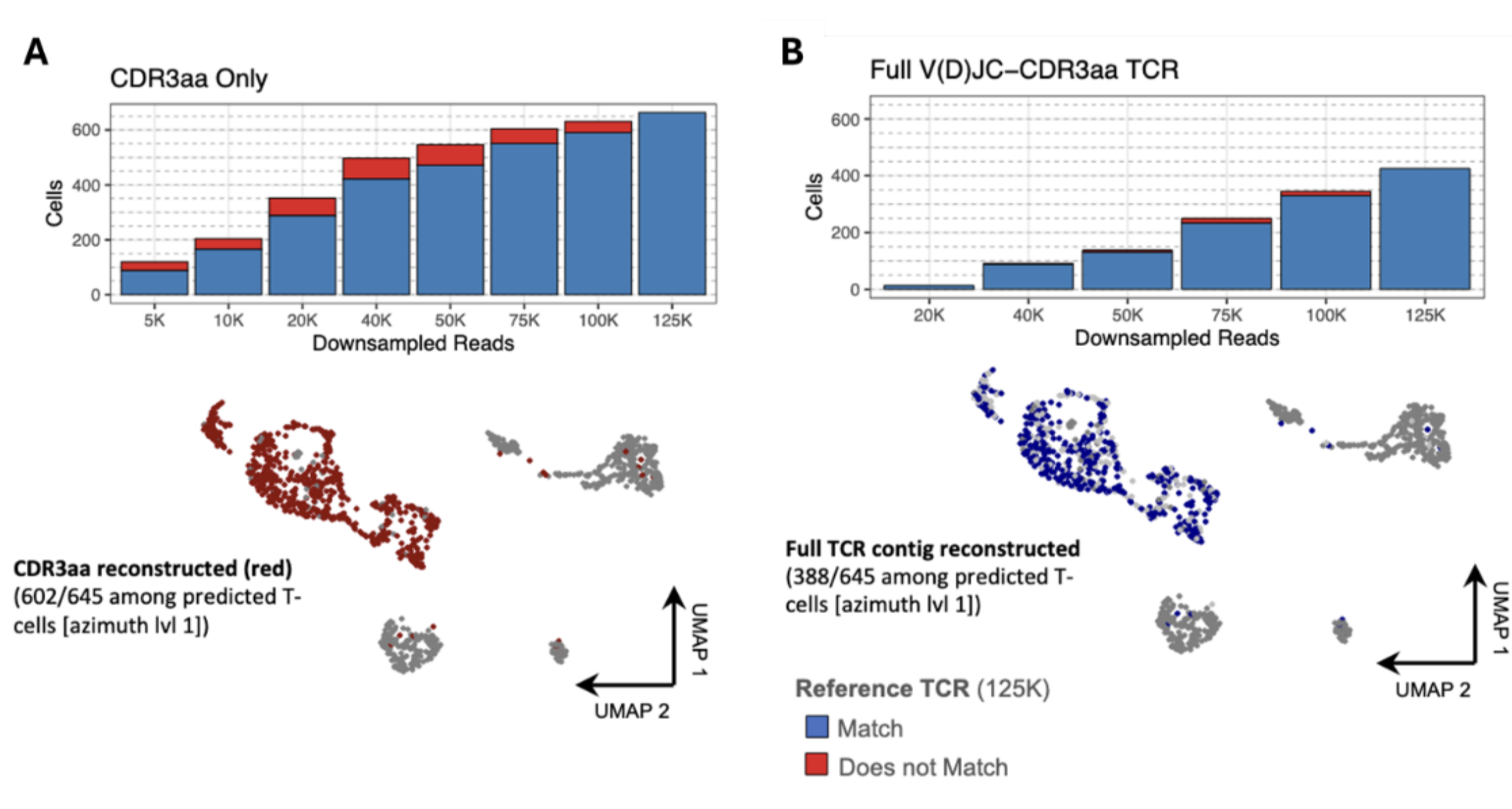
Figure 5. FLASH-seq can accurately infer CDR3aa and TCR sequences. (A) Barplot indicating the number of cells with a valid reconstructed CDR3aa sequence (TRUST4), at various downsampling depths, colored by cells with the same (blue) or different (red) CDR3aa as the 125K reference. Lower panel: UMAP visualization of the hPBMCs processed with FLASH-seq and FLASH-seq Low-Amplification, using 125K downsampled raw reads, colored by the reconstruction of the CDR3aa sequence using TRUST4. (B) Same as (A) but for reconstruction of the full V(D)JC-CDR3aa sequence. Figure modified from (4).
Hahaut and colleagues also designed FLASH-seq with automation in mind, making it highly compatible with liquid handling robots and automated platforms in 96- and 384-well plate formats. The simplified workflow and reduced number of steps make it particularly affordable and suitable for high-throughput applications.
FLASH-seq Considerations and Use Cases
While FLASH-seq offers clear advantages in sensitivity, speed, and ease of automation compared to the Smart-seq methods, if UMIs are critical for your pipeline, Smart-seq3 integrates 5′ UMIs by default, potentially making it seem a more suitable option than a non-UMI protocol.
However, including UMI in your full-length experiments comes with multiple issues that you should carefully consider.
1. To capture the UMI information present at the 5’ end of the TSO, users must perform a partial tagmentation, resulting in libraries of 500-2000 bp (3). These long molecules bind poorly to sequencing flow cells, which can decrease sequencing power.
2. As the UMIs are only present at the 5’ end of the transcripts, only a fraction of them are recovered. This results in a typical loss of 20-30% of detected genes when solely counting UMIs, leading many researchers to rely on internal, non-UMI reads to count genes, making UMIs relatively redundant.
3. The ratio of internal versus UMI reads can theoretically be fine-tuned by decreasing the fragmentation rate of the library. However, the right ratio of UMI reads can only be inferred post-sequencing and is challenging for many researchers.
4. Independent studies have observed that adding UMIs close to the 3’ end of the TSO can result in high percentages of strand invasions, creating falsely truncated molecules and skewing gene counts (4, 6).
Overall, the benefits of including UMI in the TSO may be outweighed by the negatives, as non-UMI methods like Smart-seq2 and FLASH-seq may be sufficient.
In many instances, the user-friendly nature of FLASH-seq, along with its compatibility with standard laboratory equipment already used for Smart-seq methods, offers significant advantages.
The improved sensitivity in gene detection provided by FLASH-seq enables researchers to study rare cell types in tumor microenvironments and allows for better resolution of isoform switching during development, among other biologically relevant features.
MERCURIUS™ FLASH-seq: The Future of Sensitive, Affordable, and Rapid Full-Length Single-Cell RNA-seq
Vincent Hahaut, the creator of FLASH-seq, is now the Head of R&D at Alithea Genomics. He has led the development of MERCURUS™ FLASH-seq as a kit and service to further streamline full-length single-cell sequencing for users in academia, pharma, agritech, cosmetics, and AI-driven discovery.
Vincent and the Alithea team have now made FLASH-seq as user-friendly as possible. Optimized volumes in the MERCURIUS™ FLASH-seq kit allow users to automate or to manually pipette samples for ultimate flexibility. The team has also developed a non-toxic tagmentation buffer that eliminates the need for dimethylsulfoxide (DMF), thereby increasing user safety.
They have included dyes to validate correct cell lysis and Tn5 inactivation and to provide convenient visual confirmation that these crucial stages are performing as expected. Further refinements now allow the protocol to detect even the rarest cell types from heterogeneous populations, and we provide a low RNA input version suitable for detecting 1 pg to 1 ng of RNA. MERCURIUS™ FLASH-seq yields high-quality and reproducible data from any FACS-sorted cell type that contains polyadenylated RNA, including PATCH-seq and subcellular biopsies.
Almost 10 years after Simone Picelli’s original Smart-seq2 publication, MERCURIUS™ FLASH-seq represents a much-needed improvement in sensitivity, speed, and affordability for researchers in every field or industry.
A recent independent comparative study of eight single-cell RNA-seq methods, including FLASH-seq and Smart-seq3, from the Genomics Core Facility, Erasmus University Medical Center Rotterdam, put it best (7): “While most methods are comparable in many regards, FLASH-seq and VASA-seq yielded the best metrics, e.g., in number of features.” They continued: “In general, more recently developed methods perform better. This also leads to the conclusion that older methods should be phased out.”
Still using Smart-seq2 or Smart-seq3? Try MERCURIUS™ FLASH-seq for more sensitive, reliable, and affordable full-length single-cell RNA-seq data with minimal changes to existing equipment and workflows. Contact us to access our newly launched MERCURIUS™ FLASH-seq kits or request a free consultation for our MERCURIUS™ FLASH-seq service.
References
- Picelli S, Björklund ÅK, Faridani OR, Sagasser S, Winberg G, Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nature methods. 2013 Nov;10(11):1096-8.
- Picelli S, Faridani OR, Björklund ÅK, Winberg G, Sagasser S, Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nature protocols. 2014 Jan;9(1):171-81.
- Hagemann-Jensen M, Ziegenhain C, Chen P, Ramsköld D, Hendriks GJ, Larsson AJ, Faridani OR, Sandberg R. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nature biotechnology. 2020 Jun;38(6):708-14.
- Hahaut V, Pavlinic D, Carbone W, Schuierer S, Balmer P, Quinodoz M, Renner M,