
RASL-seq is a cost-effective, targeted, high-throughput sequencing technology developed to help researchers, biotechnology, and pharmaceutical companies streamline the discovery of potential therapeutics of tomorrow with genomics (Li et al., 2012; Li, Qiu and Fu, 2012).
In the second installment of our RASL-seq mini-series, we explore the protocol behind the technology so you can decide if it fits the bill for your next drug discovery or development pipeline, or if recent developments in sequencing library preparations may be more suitable.
Here’s the must-know info about RASL-seq, from sample requirements and probe design to a step-by-step protocol breakdown.
Sample requirements for RASL-seq
You can perform RASL-seq on 1 µg total RNA or directly on cell lysates from around 3,000 cells per well in 384-well plates after treatment with given compounds of interest.
Direct use of cell lysates provides time- and cost-saving advantages for large sample sizes or pipeline automation.
Next, appropriate probe pairs must be designed to detect the splice sites or genes of interest relevant to your study.
RASL-seq probe design
The backbone of RASL-seq is its custom probe pairs. These are designed to bind to the two adjacent exons spanning a splice junction near the 3’ end of transcripts from a panel of up to 500 genes (Fig. 1).
To ensure binding to the correct location in the genome, both probes of a pair must contain a 20-nucleotide region complementary to their targeted exon sequences.
As RASL-seq is optimized for use with Illumina genetic sequencers, one probe must contain a 3’ Illumina-specific index sequence to act as an identifier to allow multiple libraries to be pooled and sequenced together. A phosphate group on the 5’ end allows both probes to form a phosphodiester bond after ligation when both probes are bound at adjacent exons.
The other probe must have a 5’ P5 adapter to allow the library to bind and generate clusters on the sequencing flow cell.
The RASL-seq process
After the design of the probe pairs, these oligos undergo a process of annealing to their complementary mRNA molecules, selection of mRNA from total RNA, ligation of oligos, elution, and barcoding, before sequencing (Fig. 1).
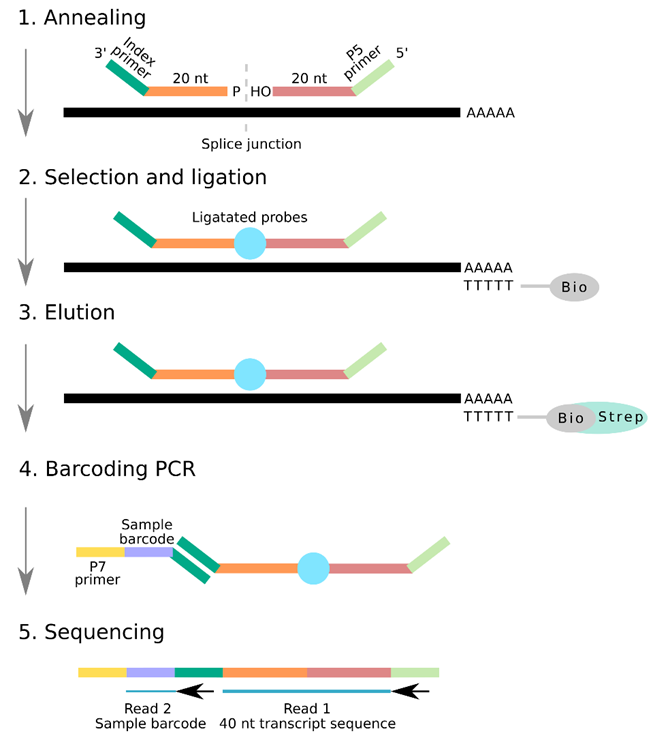
Figure 1. Schematic of the RASL-seq workflow
1. Annealing
Probe pairs anneal to the two exons adjacent to the desired splice site on a given transcript of interest.
2. Selection and ligation
mRNA transcripts are selected from total RNA by annealing to oligo(dT)-biotin beads. Addition of T4 DNA ligase ligates probe pairs when the exon-exon splice junction is present, and probes have correctly bound to their complementary templates. This generates a single PCR amplicon probe.
3. Elution
The biotin-bound mRNA strands are subsequently attached to streptavidin magnetic beads, and the ligated single probe amplicon is eluted from its mRNA template by heating.
4. Barcoding
Illumina P7 adapters attach to the 3’ index sequence, and the library undergoes PCR amplification with a set of barcoded primers. A collection of 1,536 unique barcodes allows 1,536 samples to be multiplexed in each lane.
5. Sequencing
The first read determines transcript identity by sequencing the 40-nucleotide stretch of the ligated probe from the P5 primer. The second read identifies the barcoded region from the index primer to identify which sample the transcript is from.
Other barcode-based RNA-seq methods for drug discovery
Barcoding and multiplexing strategies similar to those used in RASL-seq have been vital to advancements in modern single-cell RNA-seq approaches and recent bulk 3’ mRNA-seq technologies.
For example, MERCURIUS™ Bulk RNA Barcoding and sequencing (BRB-seq) and its RNA-extraction-free equivalent MERCURIUS™ DRUG-seq, also use a barcoding strategy but apply this to thousands of samples to simultaneously generate genome-wide transcriptional read-outs not limited to a select 500 genes.
The protocols are streamlined, automation compatible, and of a comparable price per sample to RASL-seq, making it a good option for transcriptome-wide data generation to determine the effects of therapeutics, gene perturbations, or diverse experimental conditions on gene expression.
Please contact us to learn more about RASL-seq, MERCURIUS™ BRB-seq, and MERCURIUS™ DRUG-seq.
References
- Li, H. et al. (2012) ‘Versatile pathway-centric approach based on high-throughput sequencing to anticancer drug discovery’, Proceedings of the National Academy of Sciences, 109(12), pp. 4609–4614. Available at: https://doi.org/10.1073/pnas.1200305109.
- Li, H., Qiu, J. and Fu, X.D. (2012) ‘RASL‐seq for massively parallel and quantitative analysis of gene expression’. Current protocols in molecular biology, 98(1), pp.4-13. Available at: https://doi.org/10.1002/0471142727.mb0413s98.