

A recent publication has used Bulk RNA Barcoding and sequencing (BRB-seq) to investigate one of the fundamental questions of embryonic development (Liu et al., 2020).
It provides a new hypothesis about why gene expression is more similar in the middle of embryonic development between species of the same phylum, compared to at the start or end.
In this article, we look at how BRB-seq made these findings possible and how this technology could help similar studies in the future.
Summary:
Present-day comparisons of both morphology and gene expression between species within a phylum have revealed that there is more similarity between species in the middle of embryogenesis, compared to earlier and later time-points (Kalinka et al., 2010; Irie and Kuratani, 2011). This is known as the ‘hourglass’ model and the point where species are most similar is called the phylotypic stage (Raff, 2012).
Determining the fundamental reason for this phenomenon is a major question of present-day research.
Many studies focus on the role of natural selection in this process by comparing gene expression across different species.
Here, to test a hypothesis not centered on natural selection, the authors used genetically identical Drosophila melanogaster fruit flies from identical environments to generate a transcriptomic time-course throughout embryogenesis.
Similar to previous findings in other organisms (Eling et al., 2019), they show that the hourglass pattern of gene expression is present even in genetically identical flies. The authors also suggest a novel hypothesis that this may be due in-part to the influences of histone modifications at this stage (Liu et al., 2020).
How RNA-seq was used:
Firstly, Liu et al. generated 239 high-quality transcriptomes covering eight different stages of fruit fly embryonic development using BRB-seq (Fig. 1) (Alpern et al., 2019; Liu et al., 2020).
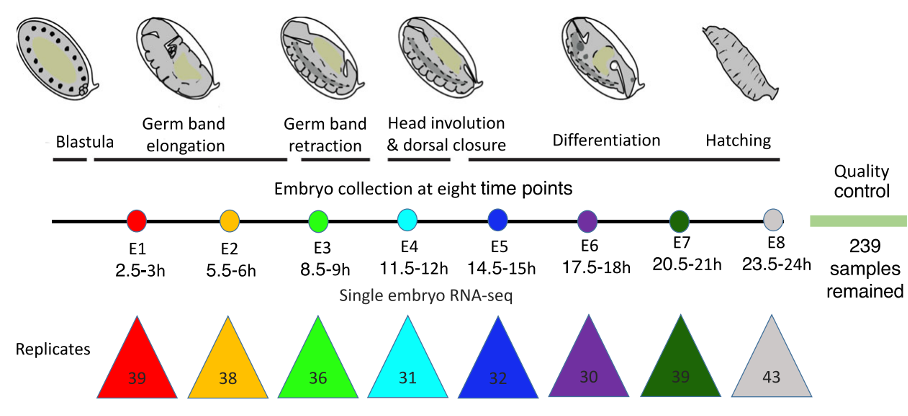
Figure 1 – Diagram of the eight stages of fruit fly embryonic development where BRB-seq was performed. The number of samples for each stage is shown. Figure modified from Liu et al., 2020.
The authors initially wanted to detect subtle natural variation in gene expression among genetically identical embryos. BRB-seq was an attractive technique due to the reduced inter-sample technical variability provided by sample multiplexing. Only four multiplexed libraries were prepared compared to 239 if Illumina TruSeq libraries were used.
Next, the authors used an adjusted standard deviation method adapted from Barroso et al., 2018 to study the variation in gene expression throughout embryogenesis. It showed that the hourglass pattern of gene expression was present even within genetically identical fly embryos. Early and late stages had significantly more variation in gene expression levels compared to the phylotypic stage three, where gene expression was more similar between samples (Fig. 2).
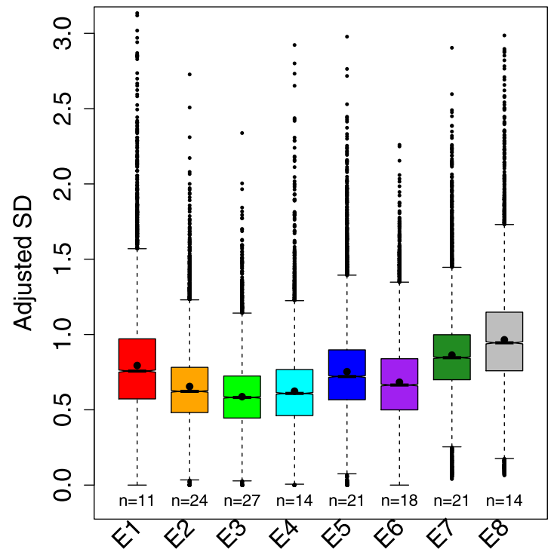 Figure 2 – Boxplots showing the variation in gene expression for the eight stages of fruit fly development. E3 is the phylotypic stage where there is the lowest variability in gene expression compared to earlier and later stages. Figure modified from Liu et al., 2020.
Figure 2 – Boxplots showing the variation in gene expression for the eight stages of fruit fly development. E3 is the phylotypic stage where there is the lowest variability in gene expression compared to earlier and later stages. Figure modified from Liu et al., 2020.
Then, the level of histone modifications over gene promoters was determined and used alongside BRB-seq gene expression data. This allowed the authors to investigate the potential mechanisms that regulate gene expression at different embryonic stages.
The combined analysis showed that histone modification signals were higher at the phylotypic stage, suggesting that histone modifications may limit gene expression variability at the mid-point in development.
How the large number of samples contributed to the results:
The initial large number of samples accounted for some eggs not being fertilized. Of 239 multiplexed BRB-seq samples, two thirds came from fertilized embryos representing eight developmental stages, but one third of all samples were removed as they were from unfertilized eggs. Between 10 to 30 samples per stage were used for further analyses.
The large number of samples for each timepoint were sufficient for sensitive pairwise Wilcoxon tests and correction for multiple testing between any two stages.
This statistical robustness would be reduced if fewer samples were used. This is especially important when looking at small changes in gene expression from genetically identical organisms.
How novel higher-throughput transcriptomics could help in similar studies:
Some novel sequencing technologies, such as BRB-seq, rely on adding a barcode to the 3’ end of mRNA molecules. This barcode allows the pooling of all samples so only one subsequent library preparation is required. This reduces both cost and technical variability between samples (Alpern et al., 2019), each of which were key considerations in this study.
For example, here the authors discarded one third of their data which would represent an enormous cost if traditional RNA-seq library preparations were used. The reduced cost of BRB-seq removes these expensive risks when working with sensitive samples and timepoints.
Furthermore, BRB-seq provides high quality data even with degraded mRNA (Alpern et al., 2019). mRNA degradation is a concern when working with low amounts of starting material. This could be critical in other developmental studies with low amounts of starting material or where material is extremely precious.
To find out more about how BRB-seq could help your study, please contact us at info@alitheagenomics.com.
References:
- Alpern, D., Gardeux, V., Russeil, J., Mangeat, B., Meireles-Filho, A.C., Breysse, R., Hacker, D. and Deplancke, B., 2019. BRB-seq: ultra-affordable high-throughput transcriptomics enabled by bulk RNA barcoding and sequencing. Genome biology, 20(1), pp.1-15.
- Barroso, G.V., Puzovic, N. and Dutheil, J.Y., 2018. The evolution of gene-specific transcriptional noise is driven by selection at the pathway level. Genetics, 208(1), pp.173-189.
- Eling, N., Morgan, M.D. and Marioni, J.C., 2019. Challenges in measuring and understanding biological noise. Nature Reviews Genetics, 20(9), pp.536-548.
- Irie, N. and Kuratani, S., 2011. Comparative transcriptome analysis reveals vertebrate phylotypic period during organogenesis. Nature communications, 2(1), pp.1-8.
- Kalinka, A.T., Varga, K.M., Gerrard, D.T., Preibisch, S., Corcoran, D.L., Jarrells, J., Ohler, U., Bergman, C.M. and Tomancak, P., 2010. Gene expression divergence recapitulates the developmental hourglass model. Nature, 468(7325), pp.811-814.
- Liu, J., Frochaux, M., Gardeux, V., Deplancke, B. and Robinson-Rechavi, M., 2020. Inter-embryo gene expression variability recapitulates the hourglass pattern of evo-devo. BMC biology, 18(1), pp.1-12.
- Raff, R.A., 2012. The shape of life: genes, development, and the evolution of animal form. University of Chicago Press.
