MERCURIUS™ formalin-fixed paraffin-embedded sequencing (FFPE-seq) is a scalable and cost-effective full-length bulk RNA sequencing technology optimized for heavily degraded RNA samples with RNA integrity numbers (RIN) as low as 1.
Despite the routine use of traditional RNA-seq in transcriptomics, standard RNA library preparation methods often require good-quality RNA of RIN ≥ 8 and generally miss the mark with their limited sample multiplexing capabilities. These limitations represent significant hurdles for large-scale studies reliant on poor-quality RNA from archival tissue stored in biobanks and preserved by fixation methods highly destructive to RNA (Zhang et al., 2017).
To unlock the potential transcriptomic discoveries hidden in the degraded RNA from these vast tissue repositories, MERCURIUS™ FFPE-seq combines the polyadenylation of degraded total RNA fragments with the power of early-stage sample barcoding. This poly(A)-tailing and barcoding strategy allows users to generate robust, reliable, and reproducible full-length transcriptome data from even the poorest quality RNA from samples fixed by any method while multiplexing up to 384 samples in the same tube.
In this article, we introduce the capabilities and workflow of MERCURIUS™ FFPE-seq and highlight how it is uniquely positioned to help your next large-scale study.
The downside of FFPE samples for RNA-seq
Tissue samples are routinely collected from patients during treatment but must be fixed to allow long-term tissue preservation and storage for their use in retrospective disease studies. Formalin-fixation of tissue followed by embedding in paraffin remains the most economical and common fixation approach, with over a billion FFPE samples stored in biobanks and hospitals worldwide (Blow 2007). Many samples are also clinically annotated with associated phenotypic data, potentially providing a rich source of clinically relevant transcriptomic information for rare conditions or various disease stages.
But, RNA extracted from FFPE specimens is of notoriously poor quality due to severe fragmentation and degradation, making it difficult to perform reliable transcriptome profiling studies with traditional RNA-seq approaches (Zhang et al., 2017). This ultimately hinders progress in the transcriptomic assessment of biomarkers, treatment responses, or disease outcomes for samples archived in biobanks globally.
Transcriptome profiling solutions for degraded RNA
To address these difficulties, an increasing number of targeted technologies such as Illumina TruSeq™ RNA Exome and TempO-Seq™ now allow gene expression profiling of degraded RNA samples but are probe-based and reliant on the hybridization of probes to specific sequences, making them unsuitable for the unbiased discovery of novel transcripts (Yeakley et al., 2017; Liu et al., 2022).
In contrast, 3’ bulk RNA-seq methods such as MERCURIUS™ DRUG-seq and BRB-seq provide unbiased gene expression information for RNA samples with low RIN values by adding barcodes and unique molecular identifiers (UMIs) to the poly(A) tail of each mRNA molecule followed by sequencing (Alpern et al., 2019). But, these methods generate reads only for the 3’ region of mRNA instead of full-length transcripts, which limits the exploration of novel isoforms, alternative splicing events, or fusion genes (Fig. 1).
To fill these gaps, MERCURIUS™ FFPE-seq now enables massively multiplexed, unbiased transcriptomics for even the poorest quality RNA from FFPE samples or any other fixation method, with high read coverage across the entire length of transcripts (Fig. 1).
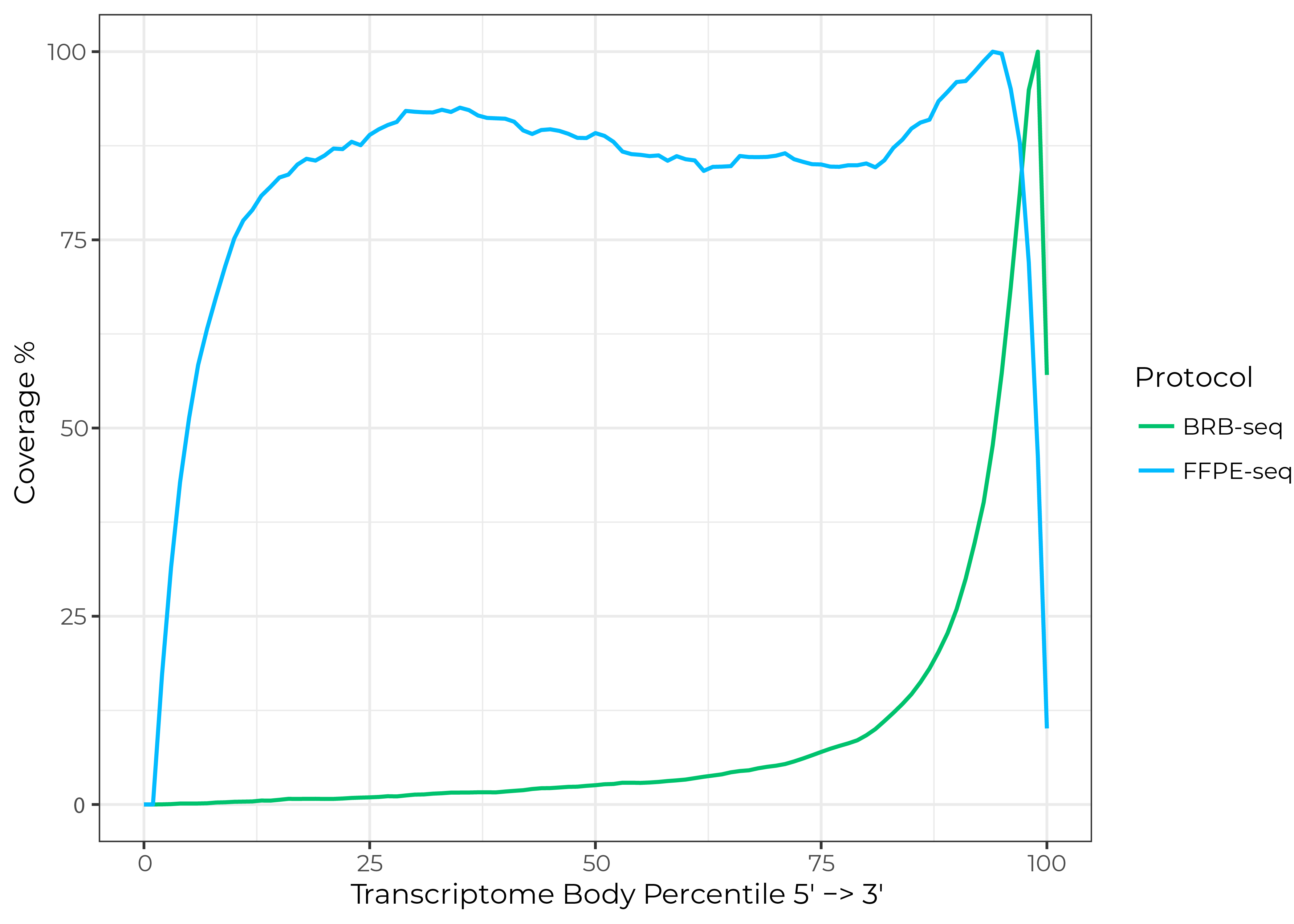
Figure 1. FFPE-seq provides read coverage across the entire length of transcripts compared to coverage only at the 3’ end with BRB-seq. Data generated from three replicates of human mixed FFPE samples sequenced at a depth of 20 million reads/sample.
The MERCURIUS™ FFPE-seq workflow
MERCURIUS™ FFPE-seq first requires users to extract total RNA from their samples. The assay is compatible with FFPE-derived RNA or RNA from any sample with RIN values as low as 1 to as high as 10. For optimal results, we recommend 500ng of total RNA per sample, and users can choose either 96 or 384-well plate formats for extensive multiplexing.
The hands-on time for 384 samples is around 3 hours and 15 minutes, with up to 6 hours and 10 minutes of incubation time.
Here’s how it works (Fig. 2):
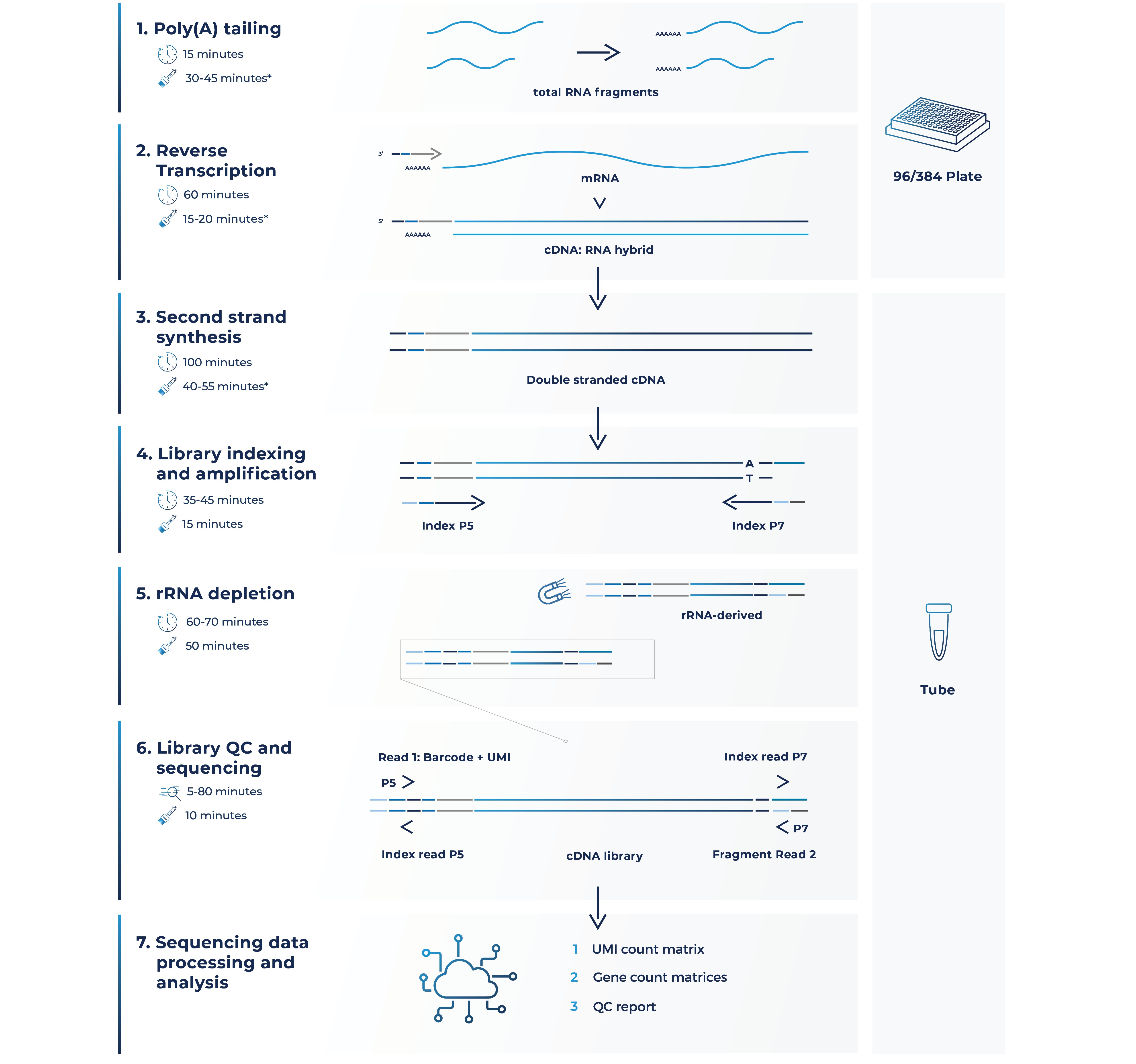
Figure 2. MERCURIUS™ FFPE-seq workflow at a glance.
1. Poly(A) tailing
Similar to other MERCURIUS™ technologies, MERCURIUS™ FFPE-seq uses highly optimized barcoded oligo(dT) primers to uniquely tag the poly(A) tail of each individual RNA molecule per sample during the first strand synthesis stage of cDNA library preparation (Alpern et al., 2019).
But, heavily degraded RNA is fragmented, with most pieces lacking the intact poly(A) tails required for barcodes to bind and prime reverse transcription.
Therefore, initial end repair and poly(A) tailing reactions are performed on each sample in 96 or 384-well plates so that each fragment of total RNA can be barcoded and primed for reverse transcription, ensuring read coverage across the full-length of transcripts regardless of RIN values (Fig. 2).
2. Barcoded reverse transcription
Next, poly(A) tails bind to oligo(dT) barcoded nucleotide sequences containing an adapter for primer annealing, a barcode that assigns a unique identifier to each RNA sample per well, and a UMI to tag each RNA fragment to distinguish between original RNA transcripts and duplicates from amplification. First-strand cDNA synthesis via reverse transcription then results in barcoded cDNA:RNA hybrids (Fig. 2).
Because the cDNA:RNA hybrids are individually barcoded per well, all samples can be pooled into the same tube from this point onwards, significantly reducing hands-on time, technical error, and consumable use.
3. Second-strand synthesis
The single tube containing the pooled double-stranded cDNA from 96 or 384 samples then undergoes second-strand synthesis (Fig. 2).
4. Library indexing and amplification
The resulting cDNA library is then indexed and amplified using a unique dual indexing approach to minimize the risk of barcode misassignment after sequencing with an Illumina sequencing platform (Fig. 2).
5. rRNA depletion and re-amplification
Ribosomal RNA (rRNA) comprises up to 80-90% of molecules in a total RNA sample, so these must be removed by rRNA depletion methods to ensure that sequencing focuses on more informative parts of the transcriptome (O’Neil, Glowatz, and Schlumpberger 2013).
After the removal of rRNA-derived cDNA, each library pool is re-amplified instead of each sample individually.
6. Library QC and sequencing
Libraries then undergo a series of quality control checks and paired-end Illumina sequencing is performed with a recommended sequencing depth between 10 to 20 million reads per sample.
7. Data processing
Samples are demultiplexed depending on the unique sample barcode detected by read 1. Read 1 also detects cDNA duplicates from amplification by indicating the UMI, while the sequence of each fragment is detected by read 2 (Fig. 2).
Finally, the generated data is aligned to the genome of choice, with QC reports and ready-to-use gene count matrices provided for downstream analysis.
Overall, the discovery of diagnostic and prognostic gene expression biomarkers will undoubtedly rely heavily on retrospective studies using historical FFPE samples or precious clinical samples with suboptimal RNA quality.
MERCURIUS™ FFPE-seq now allows researchers to access this treasure trove of biological information by providing massively multiplexed transcriptomics for entire transcripts in even heavily degraded RNA samples.
Don’t hesitate to get in touch with us to find out more about how MERCURIUS™ FFPE-seq can help your subsequent study.
References
- Alpern, D. et al. (2019) ‘BRB-seq: Ultra-affordable high-throughput transcriptomics enabled by bulk RNA barcoding and sequencing’, Genome Biology, 20(1), pp.1-15. Available at: https://doi.org/10.1186/s13059-019-1671-x.
- Blow, N. (2007) ‘Tissue issues’, Nature, 448(7156), pp.959-960. Available at: https://doi.org/10.1038/448959a.
- Liu, Y. et al. (2022) ‘Quality control recommendations for RNASeq using FFPE samples based on pre-sequencing lab metrics and post-sequencing bioinformatics metrics’, BMC Medical Genomics, 15(1), p.195. Available at: https://doi.org/10.1186/s12920-022-01355-0.
- O’Neil, D., Glowatz, H. and Schlumpberger, M. (2013) ‘Ribosomal RNA depletion for efficient use of RNA‐seq capacity’, Current Protocols in Molecular Biology, 103(1), pp.4-19. Available at: https://doi.org/10.1002/0471142727.mb0419s103.
- Yeakley, J.M. et al. (2017) ‘A trichostatin A expression signature identified by TempO-Seq targeted whole transcriptome profiling’, PloS One, 12(5), p.e0178302. Available at: https://doi.org/10.1371/journal.pone.0178302.
- Zhang, P. et al. (2017) ‘The utilization of formalin fixed-paraffin-embedded specimens in high throughput genomic studies’, International Journal of Genomics, 2017. Available at: https://doi.org/10.1155/2017/1926304.