
Shallow sequencing of MERCURIUS™ BRB-seq and MERCURIUS™ DRUG-seq library preparations is a quality control (QC) step to ensure that each multiplexed sample will obtain a uniform number of reads to generate reliable and robust data when the libraries are sequenced deeper.
Shallow sequencing with around 100,000 reads per sample ensures an equal distribution of reads per sample in the library before high-depth runs of approximately 1 to 5 million reads per sample. It does this by assessing key metrics of multiplexed libraries, such as read depth and mapping rates per sample.
Read on to find out what this quality control step is, why it is important, and if it is appropriate for your samples.
What is shallow sequencing quality control?
MERCURIUS™ BRB-seq and MERCURIUS™ DRUG-seq protocols allow up to 384 samples to be pooled in the same tube early in the library preparation workflow. MERCURIUS™ DRUG-seq differs from MERCURIUS™ BRB-seq in that it is an RNA-extraction-free protocol.
These technologies rely on unique sample barcodes. These are added to the 3’ poly-A tail of each mRNA in every sample at the reverse transcription stage during first-strand synthesis.
The resulting tagged cDNAs are then pooled in the same tube for extensive sample multiplexing.
Shallow sequencing QC uses the sample barcodes that mark each sequencing read to check key library metrics such as read depth and mapping rate per multiplexed sample. This informs on sample balancing in a library before deeper sequencing.
For libraries with varying numbers of reads per sample, balancing involves modifying the quantity of barcoded cDNA from each outlier sample to create a final library that is evenly distributed. This final library can then be used for high-depth sequencing.
Therefore, proper balancing of samples in a library allows for more uniformity across reads per sample and helps to improve the distribution of sequencing reads in the library. This further translates into more consistent sequencing results, reliable detection of gene expression, simplified data analysis and ultimately, generation of accurate biological insight.
Consequences of imbalanced libraries
If samples are not uniform or fall below our recommended quantity, quality, and purity thresholds (see below), libraries can suffer from low cDNA yields and reduced coverage for the affected samples during deeper sequencing
Conversely, a sample may obtain too many reads compared to other samples in the library if it has more starting RNA. This can reduce the amount of information obtained for samples with lower sequencing coverage in the library. Pooling together in the same library very different sample types, such as those obtained from different species or tissue types (i.e. human and mouse, or brain and liver), can also contribute to these differences.
This results in a more imbalanced distribution of reads across the multiplexed samples in both cases.
In the standard data processing pipeline the resulting raw read counts data is computationally normalized by the total amount of reads acquired for each sample as indicated by the sample barcode. This enables to perform a comparison between samples in the downstream analyses.
Importantly, this normalization results in reliable data when the amount of reads per sample is relatively uniform. Lower coverage may result in the detection of much fewer genes, making it impossible to use such samples in a comparison. Therefore in some instances, removal of the data corresponding to such samples from the library may be recommended.
Shallow sequencing QC, therefore, gives a reliable indication of any samples in the library that may be outliers in the amount of sequencing reads they would acquire in deeper sequencing. Equilibrating libraries ultimately translates into better comparisons between samples during data analysis.
Sample prerequisites
For MERCURIUS™ BRB-seq, RNA samples should be of a uniform quantity, quality, and purity to ensure an equal distribution of reads per sample in the library. These factors can affect how much cDNA is generated and, consequently, the number of reads gained per sample during sequencing.
We recommend using 10 – 1000 ng of total purified RNA per sample, with two considerations: (i) the total RNA amount per pool should be at least 1000 ng, and (ii) the minimum number of samples that can be pooled is eight. Nevertheless, the samples should always be diluted to the same RNA concentrations in all wells. RNA quality should also be as uniform as possible, with an RNA integrity number (RIN) of at least 6. Samples should also have a similar purity, with a 260/230 ratio greater than 1.5 when measured with a nanodrop indicating limited residual contamination with organic solvents from RNA extraction procedures.
As MERCURIUS™ DRUG-seq is RNA-extraction free and barcoding is performed directly on cell lysates, cell number is a proxy for RNA amount. Therefore, all wells should have similar numbers of cells.
How does shallow sequencing QC work?
We use a straightforward, three-step process for shallow sequencing QC.
1. Shallow sequencing
We sequence the library containing multiplexed barcoded samples at a depth ranging between 10,000 and 100,000 reads per sample. For each sample, only a fraction of the reverse transcribed barcoded cDNA is used for shallow sequencing (i.e., 5 µl from 20 µl per sample). Shallow sequencing gives a good approximation of what to expect from deep sequencing.
2. Data analysis to generate shallow sequencing QC metrics
We then analyze the shallow sequencing data using our QC analysis pipeline.
This generates key metrics such as rates of read mapping to exons, demultiplexing, sample read distribution, and detection of gene expression.
The shallow sequencing data is first demultiplexed using the unique sample barcodes.
We then determine the distribution of reads across all samples and identify any outlier samples with a higher or lower proportion of reads than necessary for a balanced library which may require adjustment.
The expected level of mean proportion of reads for 96 samples is 1.04%. In the example below, before the adjustment of MERCURIUS™ BRB-seq samples from a 96-well plate, some samples are under or overrepresented in the library (Fig. 1A & B). The most underrepresented sample has only 0.17% of all reads in the library, whereas the most overrepresented sample gained 2.13%, a 12.5-fold difference in read distributions (Fig. 1A & B).
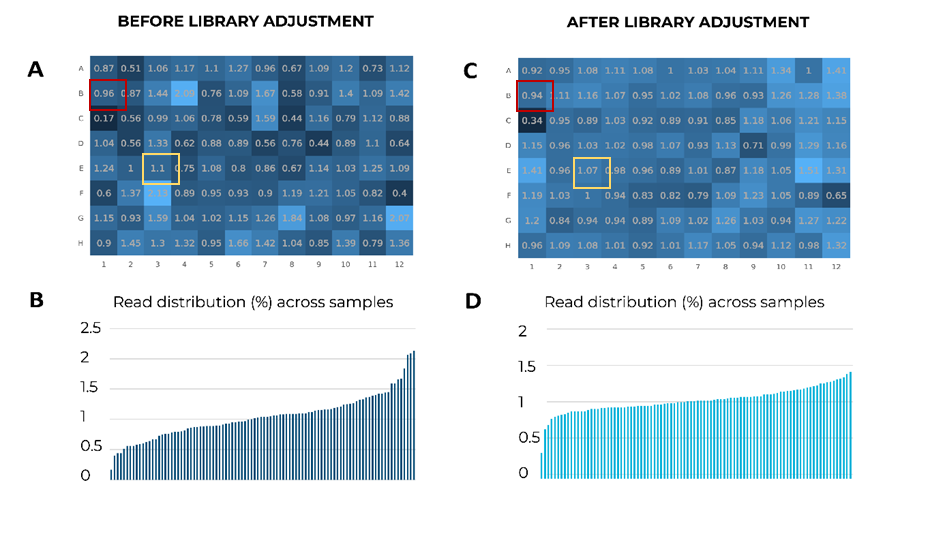
Figure 1. Comparison of samples’ read distribution before and after the adjustment from the cDNA plate, with the help of shallow sequencing. (A) Percentages of reads per sample in a 96-well plate after shallow sequencing before adjustment. There is a 12.5–fold difference between the lowest sample (red square, 0.17%) and the highest sample (yellow square, 2.13%). (B) Bar plot indicating the distribution of reads across samples after shallow sequencing before adjustment. The read distribution between samples is uneven. (C) Percentages of reads per sample in a 96-well plate after adjustment. The difference between the lowest and highest samples is reduced to a 4.4-fold. (D) Bar plot indicating the distribution of reads across samples after adjustment. The read distribution between samples is relatively homogenous.
Importantly, after libraries were generated with adjusted amounts of cDNA from the under and overrepresented samples, the difference between read distributions dropped to 4.4-fold, indicating a more homogenous read coverage per sample (Fig. 1C & D).
While the results are not identical between shallow and deeper sequencing, the level of similarity between the two can comfortably alleviate any concerns or uncertainties with the library preparation or efficacy of globin depletion.
For example, in shallow sequencing of a MERCURIUS™ Blood BRB-seq library with 48 samples, 82.8% of all genes detected were genes other than globin genes (Fig. 2A), indicating that the efficiency of globin depletion varied across the 48 samples (Fig. 2B).
Similarly, in deeper sequencing of the same MERCURIUS™ Blood BRB-seq library, 82.3% of all genes detected were non-globin genes. Each sample had similar percentages of reads mapping to genes other than globin genes as in shallow sequencing (Fig. 2C & D).
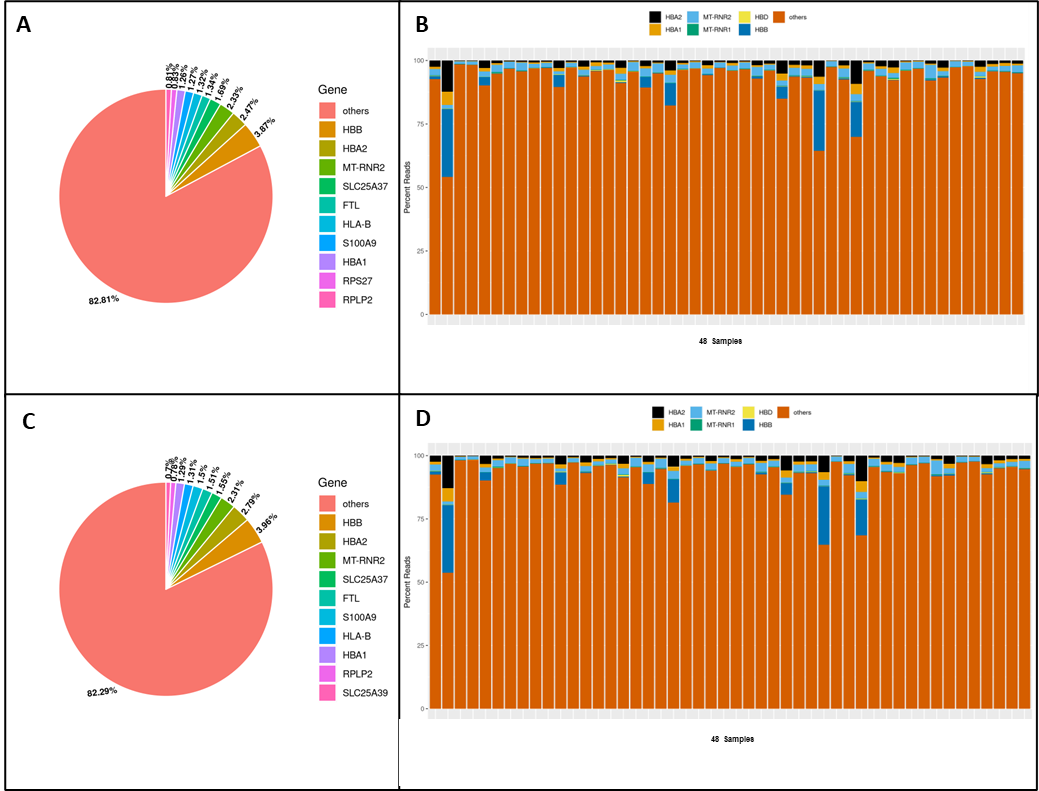
Figure 2. Comparison between shallow sequencing (0.15 million reads/sample) and deep sequencing (5.5 million reads/sample) after globin depletion with the MERCURIUS™ Blood BRB-seq service. (A) Pie Chart indicating the most expressed genes across samples after shallow sequencing. Globin genes are depleted below 10% of all detected genes. (B) Bar plot showing the depletion of globin genes per sample after shallow sequencing. (C) Pie Chart indicating the most expressed genes across samples after shallow sequencing. (D) Bar plot indicating the depletion of globin genes per sample after shallow sequencing.
3. Pass or fail decision
The above metrics help us advise on whether your libraries are suitable for deeper sequencing or if further library optimization is needed.
There are two main criteria for a pass of shallow sequencing QC:
- The distribution of reads per sample should vary by no more than fivefold between samples.
- More than 50% of all reads in a human sample should map to exons in the human genome. This 50% cutoff depends on the species investigated.
When both criteria are met, no additional library prep is necessary, and we can proceed directly with deeper sequencing.
If the libraries fail QC, we advise additional library preparation and adjustments before deeper sequencing. Libraries will then meet the library quality requirements with more homogenous read coverage per sample.
What happens if my library fails shallow sequencing QC?
If your library preparation has failed our shallow sequencing QC, we will re-prepare the library from the barcoded cDNAs. Usually, no additional reverse transcription or barcoding steps are required.
We adjust the amount of cDNA added to the new library for any samples with too many or too few reads based on the results of the shallow sequencing QC.
This ensures that all multiplexed samples have a more uniform read distribution upon deeper sequencing (Fig. 2).
When should I use it?
Alithea Genomics recommends shallow sequencing QC if this is the first time you use MERCURIUS™ BRB-seq or MERCURIUS™ DRUG-seq or if you have unfamiliar sample types (organoids, tissues, etc.).
Shallow sequencing QC is mandatory for MERCURIUS™ Blood BRB-seq as it allows us to check the efficiency of the globin depletion step.
It is also necessary where samples have variable qualities, quantities, or purities (see Sample prerequisites section) as differences in these may lead to sample over or underrepresentation in the library pool.
What is the drawback?
The main drawback of shallow sequencing QC is that it may extend the turnaround time of the BRB-seq and DRUG-seq workflows.
This depends on the outcome of the QC. For example, if the library passes QC and does not need to be re-adjusted, it will add an extra week.
However, if libraries need to be re-adjusted, it will take approximately an extra two weeks until data is ready for the final gene expression analysis stage.
There is also a small additional cost to perform the shallow sequencing QC.
The Alithea Genomics service team will provide their expertise and support to help you decide if shallow sequencing is appropriate for your samples and to maximize the success of your experiment.
Summary
Overall, the shallow sequencing QC stage of the MERCURIUS™ BRB-seq and DRUG-seq pipelines gives you peace of mind that your library preparations are balanced and suitable for deeper sequencing.
It provides confidence in library preparations where samples have variable qualities or quantities of input RNA or where researchers have unfamiliar sample types or are new to the library preparation protocol.
We provide full advice and support for any questions you may have about the library preparation and the shallow sequencing QC stages or any other stage of the pipeline.
You can contact us with any queries or ask to talk to one of our experts..