
As a researcher performing large-scale drug-discovery, genetic perturbation, or toxicogenomic screens, probe-based targeted gene expression profiling assays like Templated Oligo assay with Sequencing (TempO-seq™) from BioSpyder™ are often a suitable choice thanks to their high throughput, targeted, and relatively cost-effective nature compared to traditional RNA-seq approaches.
One area where TempO-seq™ is now gaining traction is in environmental toxicology studies, where its implementation in various international projects like the EU-ToxRisk project aims to “drive a paradigm shift in toxicology towards an animal-free, mechanism-based integrated approach to chemical safety assessment.”
Let’s dive in to see how researchers recently used TempO-seq™ to map response variability in a personalized, large-scale toxicology study, and how other more unbiased, non-targeted approaches like MERCURIUS™ DRUG-seq could provide richer transcriptome-wide data for future studies.
Personalized toxicology with TempO-seq™
Large-scale perturbation screens benefit from testing vast amounts of samples, experimental conditions, or replicates to obtain accurate, reproducible results, preferably at the lowest cost per sample without compromising data quality.
For instance, an international team of researchers from Europe and the USA recently used TempO-seq™ to quantify the interindividual variability in expression of stress response genes after treatment with broad concentrations of four different drug compounds on primary human hepatocyte (PHH) cells from 50 individuals (Niemeijer et al., 2024). They measured gene expression changes at eight and 24 hours post-exposure for a total of 8000 samples (Fig. 1).
The throughput of TempO-seq™ allowed the researchers to overcome a bottleneck of traditional toxicological assessments that often overlook the spectrum of individual patient responses, potentially underestimating risks for sensitive subpopulations.
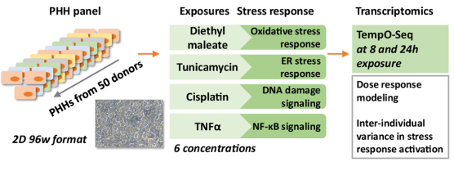
Figure 1. Schematic representation of experimental screening setup. Image taken from Niemeijer et al., 2024.
Targeted sequencing is more time-efficient compared to standard RNA-seq
Transcriptional profiling of this many samples would have been cost-prohibitive with traditional RNA-seq approaches, such as Illumina TruSeq, because RNA from each sample must be extracted individually, followed by more costly and time-consuming library preparation stages.
Instead, TempO-seq™ is RNA-extraction-free and allows extensive sample multiplexing to while reducing costs (Yeakley et al., 2017). The technology uses defined probe panels designed to annotated transcriptomic sequences.
Missed information with targeted sequencing?
While this use of defined probe sets makes for scalable and relatively cost-effective studies, the reliance on known targets comes with some issues, as the probes fundamental to TempO-seq™ are designed based upon prior knowledge of transcript sequences.
This requirement for prior knowledge of targets means that “off the shelf” probe sets available for TempO-seq™ can’t detect novel, uncharacterized, or poorly annotated transcripts unless explicitly included as custom probes in a bespoke assay. While custom assays are possible, they require meticulous probe sequence selection, slowing pipelines and increasing the expense required.
For studies like this, which focus on a particular pathway already included in the probes, targeted sequencing is likely an acceptable choice thanks to considerably lower costs for large sample numbers. Whole transcriptome sequencing would add an unnecessary amount of expression information for genes not relevant to their research question at increased expense. However, failure to obtain transcriptome-wide data could also lead to crucially missed expression information if performing a hypothesis discovery experiment.
For instance, one issue the researchers faced was that PHH cells showed dedifferentiation in cell culture, so they initially had to assess this variability between different individuals. To do this, they used TempO-seq™’s human whole-transcriptome targeted gene panel for eight different individuals. As all genes were included, the panel provided the necessary expression information to allow the researchers to adjust their cell culture conditions to limit dedifferentiation.
The more samples, the better
Overall, upon compound treatment with tunicamycin for the unfolded protein response, diethyl maleate for the oxidative stress response, cisplatin for the DNA damage response, or tumor necrosis factor alpha (TNFα) for NF-κB signaling, the researchers used the large sample size to discover that there was considerable variation in the stress response across individuals depending on the compound tested.
Crucially, smaller sample sizes tended to underestimate variability, showing exactly why high-throughput transcriptomic technologies like TempO-seq™ are crucial to enabling the inclusion of large, diverse donor panels for accurate risk assessment.
Could unbiased, transcriptome-wide approaches be a better choice?
In this study, the researchers used a targeted panel of only around 1500+ genes to assess their 8000 samples. The S1500+ panel is similar to the L1000 technology we’ve covered before, as it measures smaller sets of genes that allow users to computationally infer the expression levels of genes not directly assessed.
This gene panel was previously selected by the National Institute of Environmental Health Sciences (NIEHS) as it was determined to be:
- Representative of biological diversity
- Capable of serving as a proxy for expression changes in unmeasured genes
- Sufficient to provide coverage of well-described biological pathways
While this successfully provided insightful information as discussed, the researchers still had to employ a whole transcriptome panel for eight samples to inform on experimental optimization of culture conditions.
A “one-stop shop” technology that doesn’t require targeted panels, and instead uses ultra-high-throughput unbiased transcriptomics like MERCURIUS™ DRUG-seq, is likely a more cost-effective and efficient choice, especially for exploratory studies investigating the broad effects of perturbations with limited prior knowledge of expected transcriptomic effects.
This is especially true for early pre-clinical screening studies where the genic on- or off-target effects of a drug aren’t yet known. Unbiased read-outs of all mRNA poly(A) transcripts expressed in a sample or affected by a treatment are key pieces of information required for successful hit prioritization and core decision making without reliance on prior assumptions. MERCURIUS™ DRUG-seq provides this information by enabling comprehensive transcriptomic profiling of all transcripts, including those not captured by predefined probe panels, to help researchers avoid any blind spots in their screening data.
Scalable data generation at a low cost
Alongside its true transcriptome-wide reach, MERCURIUS™ DRUG-seq is extremely cost-effective due to its massively multiplexed nature and streamlined “hands-off” workflow that generates unbiased 3’ cDNA libraries for all poly(A) mRNA molecules in a sample.
Both TempO-seq™ and MERCURIUS™ DRUG-seq are RNA-extraction-free methods, removing costly and time-consuming RNA extraction stages; however, MERCURIUS™ DRUG-seq has the advantage that researchers can multiplex up to 384 samples in one tube very early on in the pipeline, which provides distinct experimental and cost benefits.
From the early pooling stage onwards, every sample in the single tube is treated exactly the same, minimizing the risk of technical artifacts and sample loss, that are real possibilities when processing each sample individually. This early multiplexing also drastically minimizes consumable use and hands-on time and is highly suited to automation in drug discovery pipelines.
In contrast, TempO-seq™ multiplexes samples at the end of the workflow, just before sequencing, meaning that each sample must be processed individually, boosting consumable usage and overall experimental costs despite a relatively short hands-on time. Depending on assay requirements, costs for developing a custom probe panel could also be prohibitive due to complex and time-consuming experimental setups.
Leaves nothing to chance with MERCURIUS™ DRUG-seq
Overall, probe-based gene expression profiling assays like TempO-seq™ offer targeted expression information that provides a relatively small window into the highly complex transcriptional landscape that responds to drug or genetic perturbations in large-scale studies. While this is suited to testing specific hypotheses about known gene groups or mechanistic pathways, it misses novel or unannotated transcripts while giving no information on transcript isoforms unless expensive custom-designed probe panels are used, which impact the cost and timeline of studies.
While TempO-seq™’s extensive sample multiplexing is a plus, other options like MERCURIUS™ DRUG-seq combine sample multiplexing with more cost-effective, unbiased, and truly transcriptome-wide information that ensures that you’ll leave nothing to chance in your next large-scale perturbation screen.
Schedule a call with one of our experts to find out more or head to our store to find the MERCURIUS™ product that’s right for you.
References
- Mav, D., et al. 2018. A hybrid gene selection approach to create the S1500+ targeted gene sets for use in high-throughput transcriptomics. PloS one, 13(2), p.e0191105.
- Niemeijer, M., Więcek, W., Fu, S., Huppelschoten, S., Bouwman, P., Baze, A., Parmentier, C., Richert, L., Paules, R.S., Bois, F.Y. and van de Water, B., 2024. Mapping interindividual variability of toxicodynamics using high-throughput transcriptomics and primary human hepatocytes from fifty donors. Environmental Health Perspectives, 132(3), p.037005.
- Yeakley, J.M., et al. 2017. A trichostatin A expression signature identified by TempO-Seq targeted whole transcriptome profiling. PloS one, 12(5), p.e0178302.