

DRUG-seq is a cost-effective, high-throughput bulk RNA-seq library preparation method for compound screening in drug discovery and development (Ye et al., 2018; Li et al., 2022). Digital RNA with pertUrbation of Genes (DRUG-seq) provides sensitive, unbiased gene expression readouts across the whole transcriptome and is built for the data-hungry AI-aided discovery era.
The technology is RNA-extraction-free, enabling the pharmaceutical, agritech, and cosmetics sectors to simultaneously assess the transcriptomic effects of thousands of compound treatments without modifying existing screening infrastructure, while providing unparalleled predictive power that accelerates development. The low cost, ultra-high-throughput, and scalable nature of DRUG-seq ultimately enables data-rich transcriptomic screens of larger sets of compounds under more experimental conditions than ever before, without compromising data quality compared to traditional bulk RNA-seq methods.
For industrial teams looking to perform toxicology assessment, uncover on- or off-target effects, discover biomarkers or compound mechanisms of action, triage hits faster, or generate million-sample AI-ready training datasets with robust, systems-level transcriptomics early in drug discovery pipelines, read on to learn about the DRUG-seq method and its core benefits.
DRUG-seq at a glance
DRUG-seq is commonly used in early drug discovery to generate transcriptome-wide readouts at screening scale. It is particularly suited to early hit identification, mechanism-of-action studies, and toxicology, where large numbers of compounds, doses, or conditions must be compared efficiently to optimize research spend, resources, and experimental throughput while maintaining high-content, high-throughput, and unbiased data output.
What is DRUG-seq?
In large pharmaceutical drug discovery programs, teams are under constant pressure to move from phenotypic hits to confident biological hypotheses without expanding their timelines, resources, or budgets. Traditional screening assays often lack biological breadth or depth, while conventional RNA-seq is too slow and expensive to deploy at a screening scale.
DRUG-seq emerged to fill these gaps by providing scalable transcriptome-wide readouts early enough to influence go/no-go decisions, uncover compound mechanisms of action, or identify early toxicological safety concerns crucial for faster hit triage.
However, the published DRUG-seq protocol has numerous limitations when implementing in-house, including extensive regent and workflow optimization combined with suboptimal reaction efficiency. We developed MERCURIUS™ DRUG-seq as a more robust, plug-and-play technology to help industrial sectors rapidly overcome these bottlenecks and make transcriptomic screening as easy and as efficient as possible.
Read our comparison guide to learn how MERCURIUS™ DRUG-seq, developed by Alithea Genomics, overcomes the limitations of published DRUG-seq.
MERCURIUS™ DRUG-seq’s scalability arises from early plate-based sample barcoding and multiplexing, which allow users to pool 96, 384, or 1536 samples into the same tube within a fully automated workflow designed for high-throughput screening. The technology also eliminates time-consuming RNA extraction as it’s optimized specifically for cell lysates following compound treatment. These features lead to significant reductions in cost, hands-on time,experimental variability, and consumable use while maintaining data quality and biological depth compared to traditional sample-by-sample RNA-seq methods.
Visit the MERCURIUS™ DRUG-seq technology hub to learn more about how Alithea’s optimized DRUG-seq method works for screening-scale drug discovery studies.
The DRUG-seq Workflow
The key design principle of DRUG-seq is early sample barcoding, which eliminates the need to handle each condition separately downstream.
- In the first steps of DRUG-seq, researchers culture cell lines, primary cells, or spheroids in 96, 384, or 1536-well plates, enabling hundreds to thousands of conditions to be tested in parallel. After treatment with different compounds, doses, or other perturbations, cells are lysed directly in the plate.
- Next, reverse transcription primers containing a unique sample barcode and a unique molecular identifier (UMI) tag the 3’ poly(A) tail of each mRNA molecule upon reverse transcription to cDNA via template switching.
- After barcoding and sample pooling, the multiplexed cDNA is pre-amplified by PCR and undergoes ‘tagmentation’ to add index adapters to all barcoded cDNAs. These steps follow Illumina-compatible library preparation practices, which require these indices as standard.
- Researchers then sequence the libraries and analyze the data using standard RNA-seq analysis workflows.
From this point on, each sample's cDNA is uniquely labelled with its well of origin, allowing all samples to be pooled into a single tube for simultaneous processing throughout the rest of the workflow. This significantly reduces the number of handling steps, scheduling complexity, and batch effects, while reducing plastic consumable use by up to 95%.
From a workflow perspective, this also means that experimental complexity is front-loaded into a single barcoding step, which is crucial for demultiplexing samples during final data analysis, with the UMIs enabling correction for PCR duplicates.
Data analysts generally demultiplex samples, identify UMIs for PCR duplicate correction, map reads to the genome, perform transcript counting and normalization, and conduct downstream analyses such as differential expression, unsupervised clustering, and gene ontology pathway analyses to identify mechanisms of action or cytotoxic indicators.
The uniform, large-scale, high-dimensional data is also well suited to training next-generation AI models and for pairing with high-content Cell Painting to further accelerate screening pipelines and biological discovery.
Learn how DRUG-seq captures broader biological responses than Cell Painting after treatment of HepG2 cells with 82 JUMP MoA compounds in our collaboration with Charles River Laboratories.
DRUG-seq Screening Case Study: Performance at Scale
In a representative compound screen performed in 125x 384-well plates on over 22,000 samples, 51 compounds at four doses, and six cell lines, Alithea’s MERCURIUS™ DRUG-seq detects an average of 13,000 genes at a read depth of only one million reads per sample. This high sensitivity across cell lines enables detailed pathway analysis of biological processes and compound clustering, while providing crucial systems-level biological data to determine hit leadsor early cytotoxicity (Fig. 1).
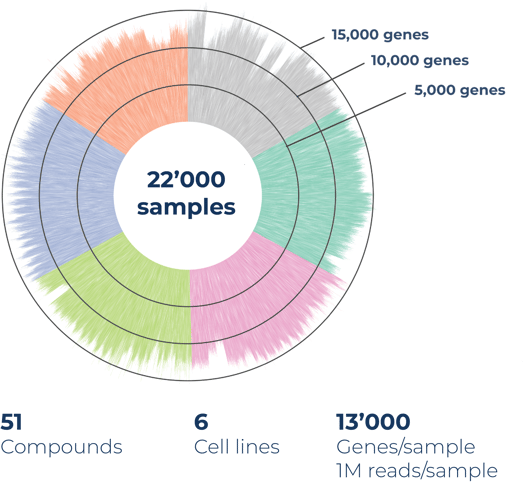
Figure 1. MERCURIUS™ DRUG-seq gene detection overview of a case study on over 22,000 samples and 51 compounds in four doses, on 6 cell lines, sequenced at 1M reads per sample.
Similarly, in a combinatorial compound screen to identify synergistic or antagonistic effects of treatment, we performed MERCURIUS™ DRUG-seq on over 11,000 samples in 36x 384-well plates across 3,840 compound combinations.
The screen detected an average of 12,000 genes per sample, even at low read depths (Fig. 2A). These large-scale, unbiased gene expression measurements enabled accurate clustering and co-clustering analyses that robustly characterized individual compounds or combinations by their transcriptomic effects (Fig. 2B).
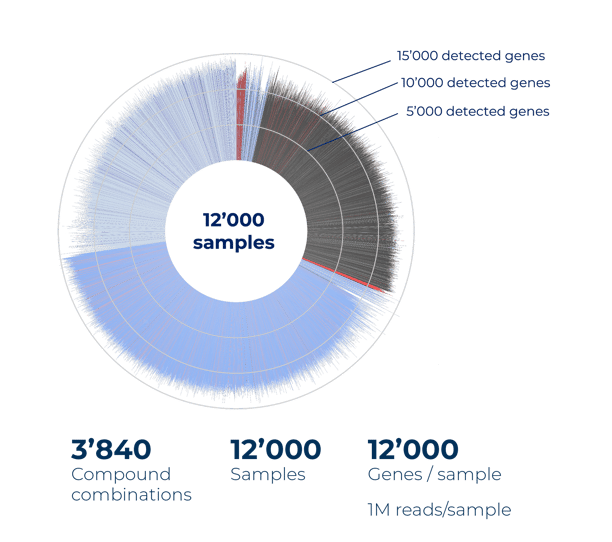
Figure 2. Combinatorial compound screen with MERCURIUS™ DRUG-seq. (A) MERCURIUS™ DRUG-seq performed on over 11,000 samples and over 3800 compound combinations detects on average 12,000 genes per sample at low read depths.
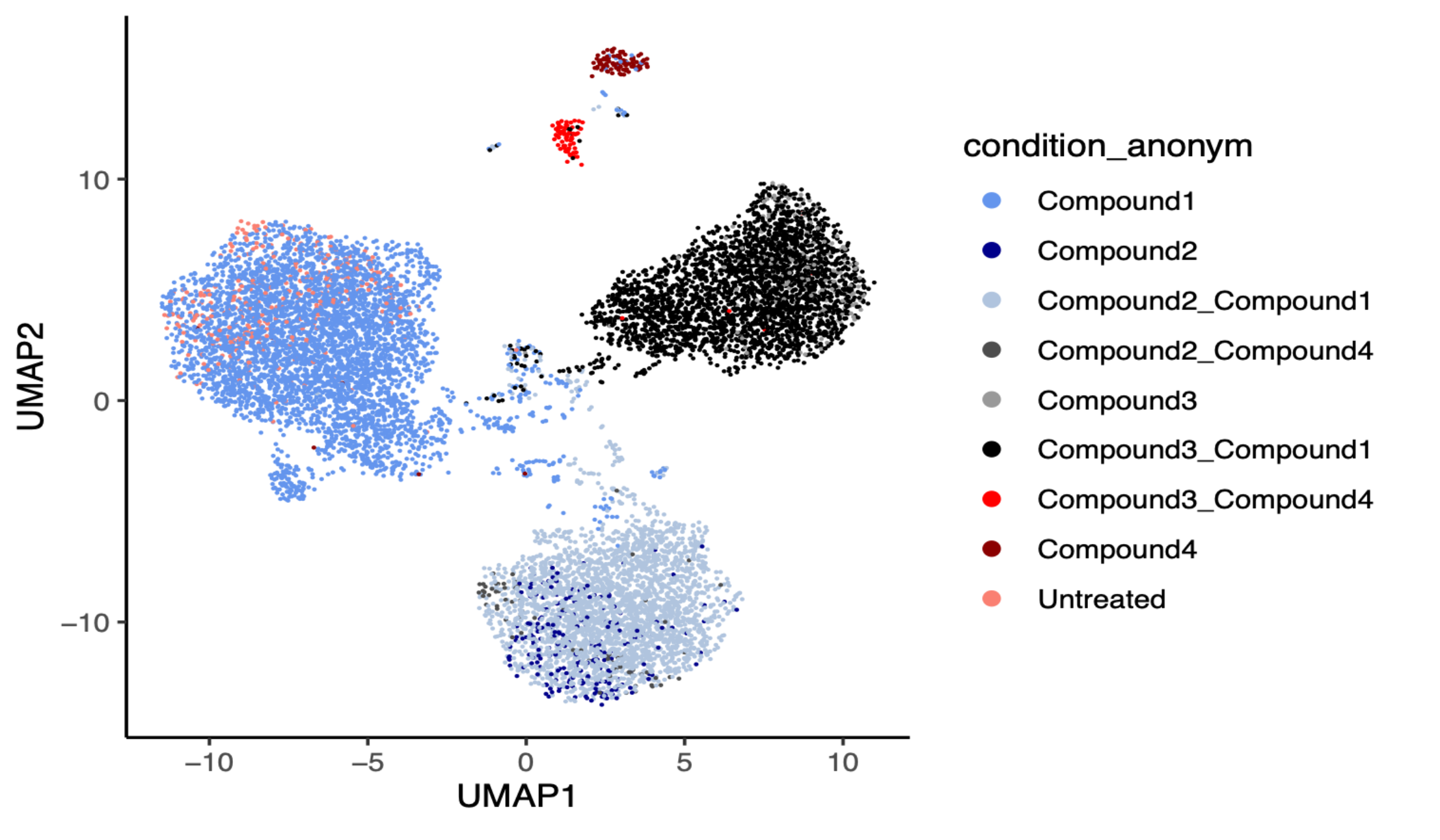
Figure 3. UMAP clustering of the MERCURIUS™ DRUG-seq data detects antagonistic and synergistic effects of different compounds based on their transcriptome profiles.
What are its benefits in drug discovery?
Because a single sequencing library is prepared directly from cell lysates for hundreds of samples simultaneously, DRUG-seq is particularly well-suited to experimental parallelization, offering significant benefits for large-scale screening pipelines and drug discovery. The ultra-scalability, combined with the high sensitivity of DRUG-seq, means it is suited to systems-level, hypothesis-generating exploration without spiraling experimental costs or pipeline planning trade-offs.
In many drug discovery programs, cost or throughput constraints mean that transcriptomic analysis is reserved for later validation stages. In contrast, DRUG-seq’s scalability and cost-effectiveness now allow researchers to generate gene expression data early enough to be considered alongside phenotypic readouts and preliminary structure–activity relationships to boost predictive power. This enables teams to incorporate pathway-level and systems-level biological context into early prioritization discussions, helping to clarify which compounds warrant further investment and which may pose biological risks.
Discover how a leading drug discovery team used MERCURIUS™ DRUG-seq to triage hits and prioritize potential therapeutic candidates for repurposing in neurological disorders.
Because DRUG-seq enables the processing and sequencing of large numbers of samples together in 96-, 384-, and 1536-sample formats, it supports direct comparison of transcriptional responses across compounds, doses, and perturbations within the same experiment. This reduces the variability associated with performing many smaller studies over time, making it easier to plan, execute, and interpret experiments while providing a better understanding of the relative biological effects across a screening campaign. For drug discovery teams, this comparability is particularly useful during hit triage, when decisions often depend on how compounds perform relative to one another rather than on absolute expression changes.
The first steps of DRUG-seq are intentionally plate-based to ensure compatibility with automated liquid-handling and compound-screening infrastructure commonly used in pharmaceutical discovery. This alignment allows integration of transcriptomic profiling into existing workflows without introducing manual bottlenecks, bespoke processing steps, or requiring expensive instrumentation. As a result, discovery teams can explore gene expression–based readouts at scale while maintaining operational consistency across screening campaigns.
For teams interested in automation, our Head of R&D shares practical insights on integrating MERCURIUS™ DRUG-seq into existing screening pipelines in a recorded webinar.
DRUG-seq Advances in 2026
One limitation of the original DRUG-seq protocol is that it detects only the 3’ region of coding mRNAs, providing robust, unbiased gene expression information but missing full-length and non-coding transcripts, as well as alternative splicing events, transcript isoforms, and fusion genes. While this is an acceptable trade-off for most studies, there’s an increasing focus on transcript isoforms and the non-coding transcriptome as therapeutic targets and indicators of mechanisms of action.
As a result, in 2025, Alithea Genomics developed MERCURIUS™ Full-Length DRUG-seq and MERCURIUS™ Total DRUG-seq, which retain the scalability and pipeline integration features of 3’ DRUG-seq but provide readouts across entire transcript lengths for the coding and non-coding transcriptome.
Similarly, drug discovery is shifting from 2D cellular models to more physiologically relevant 3D spheroid models that better recapitulate diverse cellular environments, tumor cell niches, and gene expression responses to compound treatment or other perturbations. To aid drug development teams in achieving scalable, physiologically relevant transcriptomic readouts from spheroid-based compound screens, Alithea Genomics developed MERCURIUS™ Spheroid DRUG-seq. It combines a highly optimized lysis buffer for complete spheroid lysis, with a similar barcoding strategy to 3’ DRUG-seq for efficient reverse transcription and sample multiplexing while avoiding the need for prior RNA extraction.
Frequently Asked Questions About DRUG-seq in Screening-Scale Drug Discovery
Is DRUG-seq suitable for early-stage screening, or only for validation studies?
DRUG-seq is well-suited to early-stage screening applications where large numbers of compounds, doses, or perturbations need to be compared efficiently. Its scalability and cost structure allow transcriptome-wide gene expression data to be generated early enough to inform hit triage, mechanism-of-action hypotheses, and safety signals. For later-stage validation studies requiring deeper characterization of specific transcripts or isoforms, alternative RNA-seq approaches may be more appropriate.
How does DRUG-seq differ from traditional RNA-seq?
DRUG-seq differs from traditional RNA-seq methods by offering a high-throughput, cost-efficient, and streamlined RNA-extraction-free workflow specifically designed for large-scale screening applications. One of the key differences lies in its sample multiplexing strategy, which barcodes and pools samples at the earliest step of the protocol right after cell lysis. The entire library preparation then proceeds in a single tube, rather than sample by sample as with conventional RNA-seq library preparation methods, significantly reducing both hands-on time and reagent costs.
How does DRUG-seq compare to targeted gene expression panels?
Targeted gene expression panels focus on predefined gene sets and are often used when the biological pathways of interest are already well understood. In contrast, DRUG-seq provides an unbiased, transcriptome-wide readout, making it more suitable for hypothesis-generating studies and for decision-making at the discovery stage. While targeted panels can be efficient for specific questions, they may miss unexpected transcriptional responses that influence compound prioritization.
Does shallow sequencing limit biological insight?
Shallow sequencing at 1M to 5M reads per sample provides meaningful biological insight in screening contexts where relative comparisons between conditions are more important than exhaustive transcript coverage. DRUG-seq detects around 15,000 genes at depths of below 5M reads per sample, providing gene expression patterns and differentially expressed genes at substantially lower read depths than conventional RNA-seq. The appropriate sequencingdepth depends on the research question, but deeper sequencing is not always required to support early discovery decisions.
What types of cell models are compatible with DRUG-seq?
DRUG-seq is compatible with a wide range of cell models, including most 2D immortalized cell lines and primary cells. Recent DRUG-seq variants, such as MERCURIUS™ Spheroid DRUG-seq, include an efficient lysis buffer optimized for 3D cellular models, while maintaining scalability for screening studies. As with any transcriptomic approach, sample type and quality should be considered during experimental planning.
Can DRUG-seq be integrated into existing automated screening workflows?
Yes. DRUG-seq is designed to align with standard plate-based screening and automated liquid-handling systems commonly used in industrial drug discovery. Early barcoding and pooled processing reduce manual handling steps and support integration into existing workflows without requiring bespoke instrumentation or major process changes.
When is DRUG-seq not the right approach?
DRUG-seq may not be the best choice for large-scale studies that require full-length transcript information, detailed isoform analysis, or very deep sequencing of a small number of samples. In these cases, MERCURIUS™ Full-Length or Total DRUG-seq methods may be more appropriate. Selecting the right transcriptomic approach depends on the study’s goals, scale, and the type of biological insight required at that stage of discovery.
If you’re evaluating whether transcriptome-wide screening is appropriate for your compound volume, timelines, and decision points, our team can help you assess fit and feasibility.
References
- Ye, C., Ho, D.J., Neri, M., Yang, C., Kulkarni, T., Randhawa, R., Henault, M., Mostacci, N., Farmer, P., Renner, S. and Ihry, R., 2018. DRUG-seq for miniaturized high-throughput transcriptome profiling in drug discovery. Nature communications, 9(1), pp.1-9.
- Li, J., Ho, D.J., Henault, M., Yang, C., Neri, M., Ge, R., Renner, S., Mansur, L., Lindeman, A., Kelly, B. and Tumkaya, T., 2022. DRUG-seq Provides Unbiased Biological Activity Readouts for Neuroscience Drug Discovery. ACS Chemical Biology. 17(6), pp.1401-1414


