

Novel sequencing technologies based on the 3' barcoding of mRNA now enable transcriptomic studies with higher sample numbers and lower costs than ever before.
In this article we compare two of these techniques; Single Cell RNA Barcoding and sequencing (SCRB-seq) and Bulk RNA Barcoding and sequencing (BRB-seq) (Soumillon et al., 2014; Alpern et al., 2019).
What is SCRB-seq?
SCRB-seq is a high throughput technology for the mRNA sequencing of large numbers of single cells. It is reliant on the early barcoding and subsequent multiplexing of samples (Soumillon et al., 2014).
Single cells are isolated into wells and lysed. mRNA transcripts are then annealed to custom primers comprising a poly(T) tract, unique molecular identifier (UMI) and well barcode. Template switching reverse transcription and PCR amplification generates full length, barcoded cDNA.
Because mRNA from each cell (one per well) has a unique barcode, researchers can pool all samples and identify each after sequencing. They can also use the UMI to detect PCR duplicates.
Libraries are then prepared for the multiplexed samples using the Nextera XT library prep with modified i5 primers, followed by sequencing.
The creators of SCRB-seq used it to profile over 12,000 cells throughout the development of human fat cells revealing major differences in gene expression between different cell types and time points (Soumillon et al., 2014).
Some studies have used unmodified SCRB-seq for bulk samples, however Alpern et al. optimized BRB-seq specifically for this purpose, with superior results (Cacchiarelli et al., 2015; Kilens et al., 2018; Alpern et al., 2019).
How is the BRB-seq workflow different from SCRB-seq?
Like SCRB-seq, the primer barcode used in BRB-seq includes a poly(T) tract, UMI and well barcode, but the workflow differs from SCRB-seq in several key ways (Fig. 1) (Alpern et al., 2019).
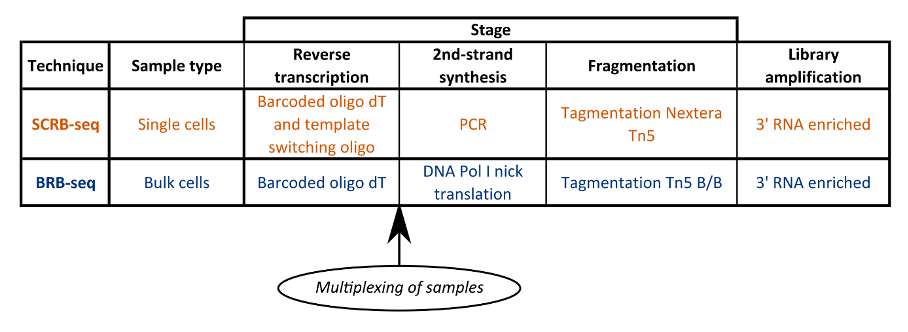
Figure 1. Table comparing SCRB-seq and BRB-seq workflows.
Firstly, as this is a bulk RNA-seq method, thousands of cells from the same experimental condition are lysed in the same well. Material is usually abundant in bulk samples, therefore the authors recommend 50-100ng of input RNA as the starting quantity if 20 samples are to be multiplexed.
Secondly, the optimized BRB-seq barcoding primers contain a poly(T) tract separated from the UMI by five random non-T nucleotides. In SCRB-seq, Alpern et al. found a bias towards reads with primers containing longer poly(T) tracts. These five additional nucleotides removed this bias and made sequencing data more diverse.
Thirdly, in BRB-seq, Alpern et al. used DNA PolI Nick translation instead of template switching during first-strand synthesis and reverse transcription by PCR. Removal of the template switch stage eliminated bias towards fully reverse transcribed molecules and potential artifacts related to strand invasion (Tang et al., 2013).
Finally, tagmentation was optimized by using a B/B tn5 transposase loaded with only i7 Illumina adapters, which increased tagging efficiency and reduced the number of PCR cycles.
Which technique is best for bulk RNA-seq?
Researchers designed SCRB-seq to profile the transcriptomes of single cells and not bulk samples.
In contrast, BRB-seq was optimized especially for bulk RNA-seq experiments. As a result, it gives much more sensitive and uniform data when compared to SCRB-seq on bulk samples (Alpern et al., 2019).
When SCRB-seq and BRB-seq were directly compared to the ‘gold standard’ Illumina TruSeq stranded mRNA method, the ratio of reads mapping to annotated genes using BRB-seq was 22% higher than for SCRB-seq and only slightly lower than TruSeq (Alpern et al., 2019).
Alpern et al. also compared the ability of SCRB-seq and BRB-seq to detect differential expression versus the TruSeq method. Strikingly, BRB-seq detected only 7% less DE genes than TruSeq, whereas SCRB-seq detected 35% less DE genes (Fig. 2) (Alpern et al., 2019).
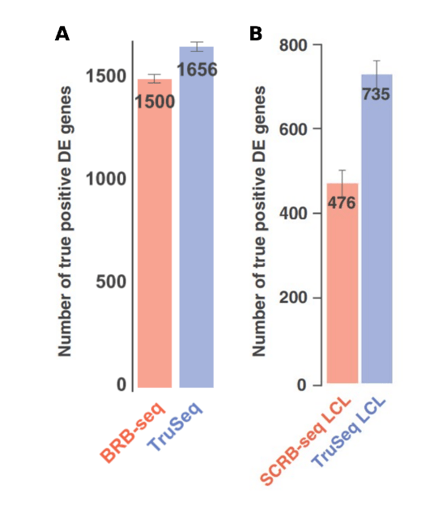 Figure 2. Evaluation of the performance of BRB-seq (A) and SCRB-seq (B) relative to TruSeq using the data downsampled to 1M single-end reads and shown by the number of “true positive” DE genes. This represents a subset of DE genes identified using the full TruSeq 30M read paired-end set. Figure modified from Alpern et al., 2019.
Figure 2. Evaluation of the performance of BRB-seq (A) and SCRB-seq (B) relative to TruSeq using the data downsampled to 1M single-end reads and shown by the number of “true positive” DE genes. This represents a subset of DE genes identified using the full TruSeq 30M read paired-end set. Figure modified from Alpern et al., 2019.
Overall, BRB-seq is a robustly benchmarked, large-scale transcriptomics approach for bulk samples that produces data of superior quality than that provided by SCRB-seq.
To find out more about how BRB-seq can be used in your study, please contact us at info@alitheagenomics.com.
References:
- Alpern, D., Gardeux, V., Russeil, J., Mangeat, B., Meireles-Filho, A.C., Breysse, R., Hacker, D. and Deplancke, B., 2019. BRB-seq: ultra-affordable high-throughput transcriptomics enabled by bulk RNA barcoding and sequencing. Genome biology, 20(1), pp.1-15.
- Cacchiarelli, D., Trapnell, C., Ziller, M.J., Soumillon, M., Cesana, M., Karnik, R., Donaghey, J., Smith, Z.D., Ratanasirintrawoot, S., Zhang, X. and Sui, S.J.H., 2015. Integrative analyses of human reprogramming reveal dynamic nature of induced pluripotency. Cell, 162(2), pp.412-424.
- Kilens, S., Meistermann, D., Moreno, D., Chariau, C., Gaignerie, A., Reignier, A., Lelièvre, Y., Casanova, M., Vallot, C., Nedellec, S. and Flippe, L., 2018. Parallel derivation of isogenic human primed and naive induced pluripotent stem cells. Nature communications, 9(1), pp.1-13.
- Soumillon, M., Cacchiarelli, D., Semrau, S., van Oudenaarden, A. and Mikkelsen, T.S., 2014. Characterization of directed differentiation by high-throughput single-cell RNA-Seq. BioRxiv, p.003236.
- Tang, D.T., Plessy, C., Salimullah, M., Suzuki, A.M., Calligaris, R., Gustincich, S. and Carninci, P., 2013. Suppression of artifacts and barcode bias in high-throughput transcriptome analyses utilizing template switching. Nucleic acids research, 41(3), pp.e44-e44.


