
Determining the transcriptomic effects of novel compounds on cells for large-scale therapeutic or toxicology screens is a critical component of drug development pipelines, allowing researchers to assess the on- or off-target effects of a drug, its toxicity, and possible mechanisms of action.
To accelerate these discoveries, the development of novel transcriptome profiling methods is progressing at breakneck speed with an eye on ever-higher sample throughput and lower costs. For drug development, this means larger screens with more experimental conditions, and more replicates, providing broader knowledge for crucial decision points.
But, with the pace of the development of transcriptomic technologies, it may be challenging to keep up to speed with which approach is most suitable for your needs.
Here we give you an insight into how three recent high-throughput transcriptome profiling technologies compare. We look at how MERCURIUS™ DRUG-seq from Alithea Genomics compares to TempO-Seq™ from BioSpyder™ and RASL-seq.
Diverse approaches to transcriptome profiling
Traditional transcriptome profiling methods such as bulk RNA-seq require expensive library preparation steps to isolate and fragment mRNA and reverse transcribe mRNA to cDNA, followed by adapter ligation and PCR amplification. Each sample must be prepared individually, increasing cost and handling time while limiting sample throughput.
Different approaches to library preparation, such as MERCURIUS™ DRUG-seq, TempO-Seq™, and RASL-seq, now address these shortcomings in traditional methods by harnessing sample barcoding to generate a single sequencing library that contains multiple distinct samples. Each technology does this in different ways (Table 1).
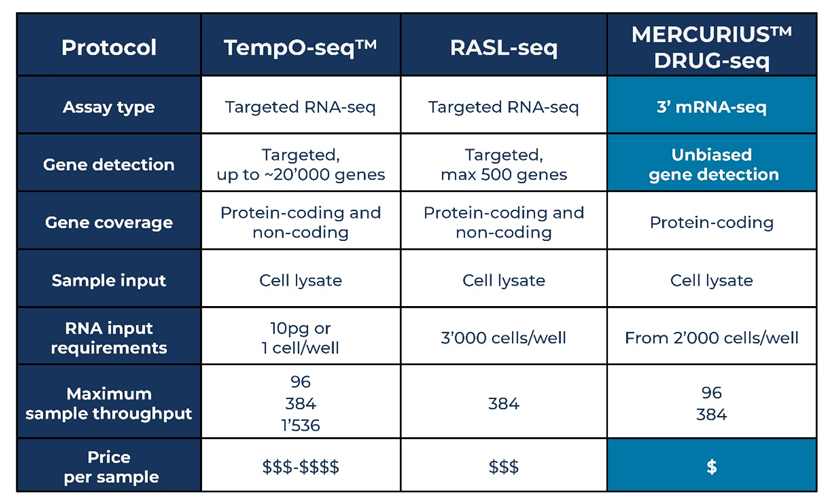
Table 1. Comparison of MERCURIUS™ DRUG-seq, TempO-Seq™ and RASL-seq
MERCURIUS™ DRUG-seq is a bulk 3’ mRNA-seq method reliant on highly optimized and rigorously evaluated sample barcodes and unique molecular identifiers to tag the 3’ poly(A) tail of all mRNA molecules during the first-strand synthesis step of cDNA library preparation. Around 20,000 genes can be detected at a sequencing depth of five million reads.
MERCURIUS™ DRUG-seq provides an unbiased window into the transcriptome as it is not reliant on probes designed to known transcripts. This means it is highly amenable to discovering novel transcripts, a layer of information missed with hybridization-based approaches such as TempO-Seq™ and RASL-seq.
TempO-Seq™ from BioSpyder™ also uses sample barcoding but combines it with dual probe hybridization and ligation where paired detector oligos target defined sequences from known sequence databases within around 20,000 transcripts (Yeakley et al., 2017).
Similarly, RASL-seq also relies on probes targeting known sequences, making it unsuitable for discovering novel transcripts, and is limited to detecting a maximum of 500 genes (Li et al., 2012).
MERCURIUS™ technology excels at multiplexing
Barcoding-based sample multiplexing now permits ultra-high-throughput screens to advance the discovery and development of potential therapeutics or to accelerate the understanding of fundamental biology or disease processes.
MERCURIUS™ DRUG-seq allows researchers to multiplex up to 384 samples in one tube for sequencing in the same run while allowing the detection of similar numbers of genes as the traditional Illumina TruSeq library preparation method at the same sequencing depth.
TempO-Seq™ allows the multiplexing of 96, 384, or 1,536 samples in the same tube, and RASL-seq allows 384 samples to be multiplexed.
Is prior RNA extraction necessary?
One key consideration for drug screening studies aiming to increase throughput while reducing cost and handling time is whether RNA must be extracted before library preparation.
MERCURIUS™ DRUG-seq uses highly optimized lysis buffers for cell lysis of 2D cell cultures and organoid models to efficiently generate library preps without prior RNA isolation.
TempO-Seq™ and RASL-seq also have flexible inputs with purified RNA, cell lysates, or formalin-fixed paraffin-embedded tissues (FFPE).
Each method lends itself to automation thanks to flexible sample inputs not reliant on prior RNA extraction and 96 or 384-well plate formats suitable for liquid handling machines.
Suitable for low amounts of starting RNA
Transcriptomic screens using human samples are often limited by low RNA quantities or poor RNA integrity due to sample acquisition, manipulation, or storage.
To permit robust results from these precious samples, it is imperative to choose a technology with RNA quality and quantity recommendations aligned to that of your samples.
MERCURIUS™ DRUG-seq requires from 2,000 cells per well, whereas TempO-Seq™ requires as little as 10 pg of RNA (around one cell’s worth) and works with degraded RNA with a RIN of 1 – 3 as is common to FFPE samples.
RASL-seq requires at least 3,000 cells per well.
The cost factor
Budgeting for a transcriptomic experiment is a key component when planning any study.
Thanks to the sample multiplexing capabilities of MERCURIUS™ DRUG-seq, TempO-Seq™ and RASL-seq, each technology is cheaper than traditional RNA-seq technologies which require expensive library preparation and deeper sequencing to gain similar amounts of gene expression information.
Out of the three technologies, MERCURIUS™ DRUG-seq is the most cost-effective method with the lowest price per sampleb (from $2 per sample) and an unbiased transcriptome-wide read-out. RASL-seq is the next lowest but information is only provided for 500 genes instead of transcriptome-wide. TempO-Seq™ provides insight into 20,000 pre-defined transcript sequences but comes in as the most expensive method.
A crucial decision
Overall, choosing a library preparation technique is an essential step for any drug discovery pipeline requiring a transcriptomic read-out. The correct choice ultimately depends on sample type, RNA amount, RNA quality, and desired multiplexing capacity.
To find out more about MERCURIUS™ DRUG-seq, please contact us.
References
-
Li, H. et al. (2012) ‘Versatile pathway-centric approach based on high-throughput sequencing to anticancer drug discovery’, Proceedings of the National Academy of Sciences, 109(12), pp. 4609–4614. Available at: https://doi.org/10.1073/pnas.1200305109.
-
Yeakley, J.M. et al. (2017) ‘A trichostatin A expression signature identified by TempO-Seq targeted whole transcriptome profiling’, PloS one, 12(5), p.e0178302. Available at: https://doi.org/10.1371/journal.pone.0178302.