
RNA-seq experiments rely on robust library prep stages to generate accurate and reliable transcriptomic data. Different library preparation technologies, such as Illumina TruSeq library prep and MERCURIUS™ BRB-seq, detect similar numbers of expressed genes even at low sequencing depth.
This article highlights how MERCURIUS™ BRB-seq combines scalability, multiplexing, and cost efficiency without compromising on data quality and gene detection.
What is Illumina TruSeq library prep?
The TruSeq stranded mRNA library prep kit from Illumina is a bulk RNA-seq method. In this method, mRNA molecules are enriched via poly-A capture, fragmented, and reverse transcribed. Sequencing adapters are then ligated to the resulting cDNA and fragments are sequenced.
Researchers assemble all the fragments from full mRNA transcripts in post-sequencing data analysis and determine their quantity to get a read-out of gene expression.
What is MERCURIUS™ BRB-seq?
MERCURIUS™ BRB-seq differs from TruSeq library prep because it is a bulk 3’ mRNA-seq technology (Alpern et al., 2019).
This means that the method adds sample barcodes and unique molecular identifiers (UMIs) to the 3’ poly-A tail of each mRNA molecule at the reverse transcription step early in the workflow, before fragmentation. Full-length, barcoded cDNA is then fragmented and indexed for sequencing.
Only the 3’ region is sequenced instead of all fragments of the mRNA transcript.
The sample barcode allows researchers to prepare hundreds of samples in the same tube. Following sequencing, these barcodes and UMIs are identified, and the amount of mRNA is accurately quantified in the data analysis stage.
This results in highly accurate transcriptomic data of a similar quality to Illumina TruSeq library prep, but at a much-reduced cost.
This is due to both sample barcoding and the reduced sequencing depth required.
What is sequencing depth and why is it important?
Sequencing depth in RNA-seq refers to the number of sequenced reads for a particular sample.
Deeper sequencing allows marginally more transcripts to be detected than at shallow depths because a larger proportion of the transcriptome can be covered. The additional genes detected are usually lowly expressed genes.
Importantly, the number of additional transcripts detected at a higher depth of sequencing is relatively small and not proportional to the number of additional reads and increased cost required (Fig. 1).
For example, both Illumina TruSeq library prep and MERCURIUS™ BRB-seq detect around 18,000 genes at a read depth of five million reads (Fig. 1). For standard transcriptomic experiments, this is likely sufficient for most pipelines. However, the number of detected genes is still comparable between the two mRNA library prep kits even at eighty million reads.
Researchers detect only around an additional 2,000 genes when sequencing depth is doubled to ten million reads (Fig. 1). These additional genes, therefore, come at a significant cost (Fig. 2).
-png.png)
Figure 1 – Graphs indicating the number of expressed genes detected with MERCURIUS™ BRB-seq or Illumina TruSeq library preps at different depths of sequencing per sample.
The cost of sequencing depth to detect lowly expressed genes
Whether the cost of detecting lowly expressed genes is worth it depends on your research question and experimental design.
At a sequencing depth of 10 million reads, the additional 2,000 genes will have a cost of 15$ per sample (Fig. 2).
For a sequencing depth of 20 million reads, the additional 4,000 genes detected will cost you approximately 30$ per sample (Fig. 2).
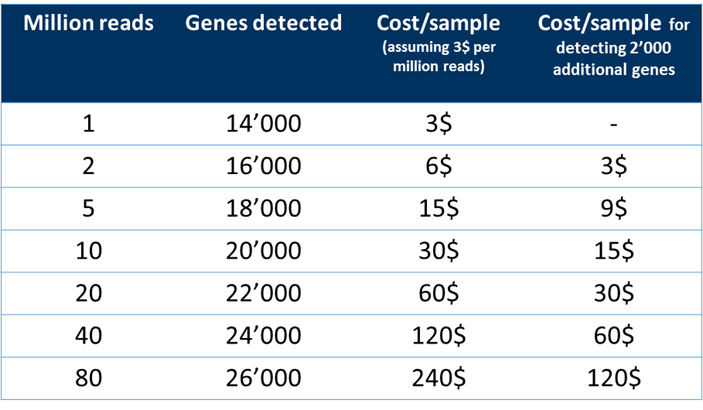
Figure 2. Table indicating the number of genes detected at different sequencing depths and the cost per sample for all and additional genes detected at higher depths.
For large-scale MERCURIUS™ BRB-seq or DRUG-seq studies with hundreds or thousands of samples, the cost to detect these lowly expressed genes quickly adds up.
It would be more cost-effective to spend this money on an increased number of biological replicates. Adding more replicates significantly enhances your power to accurately estimate absolute or differential expression levels, compared to increasing the depth of sequencing for a few samples (Liu et al., 2014).
Please contact us to find out more about MERCURIUS™ BRB-seq, MERCURIUS™ DRUG-seq, or Illumina TruSeq library preps.
References
- Alpern, D. et al. (2019) ‘BRB-seq: ultra-affordable high-throughput transcriptomics enabled by bulk RNA barcoding and sequencing’. Genome biology, 20(1), pp.1-15. Available at: https://doi.org/10.1186/s13059-019-1671-x.
- Liu, Y. et al. (2014) ‘RNA-seq differential expression studies: more sequence or more replication?’, Bioinformatics, 30(3), pp.301-304. Available at: https://doi.org/1093/bioinformatics/btt688.