Despite its widespread use, RNA-seq is still too laborious and expensive to replace RT-qPCR as the default gene expression analysis method. We anticipate that RNA-seq could become new basic laboratory practice, as a more accurate method for determination of gene expression values than microarrays and given its capacity to generate genome-wide transcriptomics information. But current workflows take too much hands-on time and are too expensive to replace qPCR practice.
Currently, the de facto standard workflow for bulk transcriptomics is the directional dUTP approach and its commercial adaptation “Illumina TruSeq Stranded mRNA”. However, like most current RNA-seq protocols, TruSeq relies on late multiplexing, which necessitates the processing of samples on a one-by-one basis. To overcome this limitation, BRB-seq uses early multiplexing to produce 3′ cDNA libraries, which provides great capacity for transforming large sets of samples into a unique sequencing library. This allows to process thousands of samples by combining the sample barcode indexing with the library indexing, which makes the design of the project very flexible. Moreover, it simplifies a lot the handling of the samples, which not only reduce the hands-on time, but also reduce cost significatively, and thus provide a much cheaper solution per sample than what TruSeq can offer.
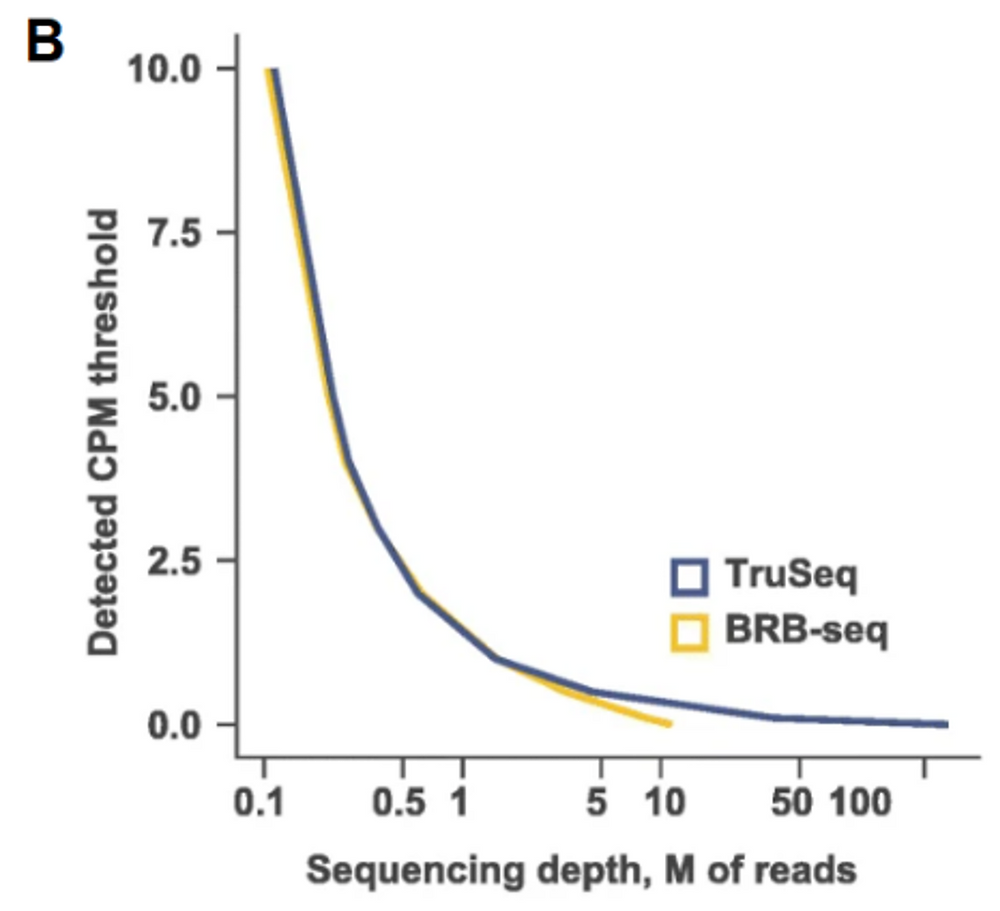
In terms of performances, like gene detection, or gene expression levels, TruSeq and BRB-seq have similar sensitivity at same sequencing depth (Fig. A, B). Both methods are stranded, which allows the assignment of reads to the correct gene strand. BRB-seq also shows greater tolerance for lower RNA quantities, RNA qualities and even works on fragmented RNA samples, which makes it the method of choice in case of low input, or degraded RNA. It also has the advantage of using Unique Molecular Identifiers (or UMIs) to unbiasedly estimate the duplicate levels in the sequenced data. This is particularly useful as a QC, but also for processing of the final data, to remove any source of noise that would come with the amplification step, which is in general not done for TruSeq libraries, because of lacking this information.
The principal limitation of BRB-seq is its incapacity to address splicing events, since it’s 3’-based. However, most transcriptomics studies do not require or exploit full transcript information, implying that standard RNA-seq methods tend to generate more information than is typically required. This unnecessarily inflates the overall experimental cost, rationalizing why 3′-end profiling approaches have already been proven effective to determine genome-wide gene expression levels. Moreover, 3’-based methods have the advantage of not requiring gene-length normalization, which is known to be biased to the gene annotation used, and thus 3’-based methods allow for more accurate determination of gene expression values.
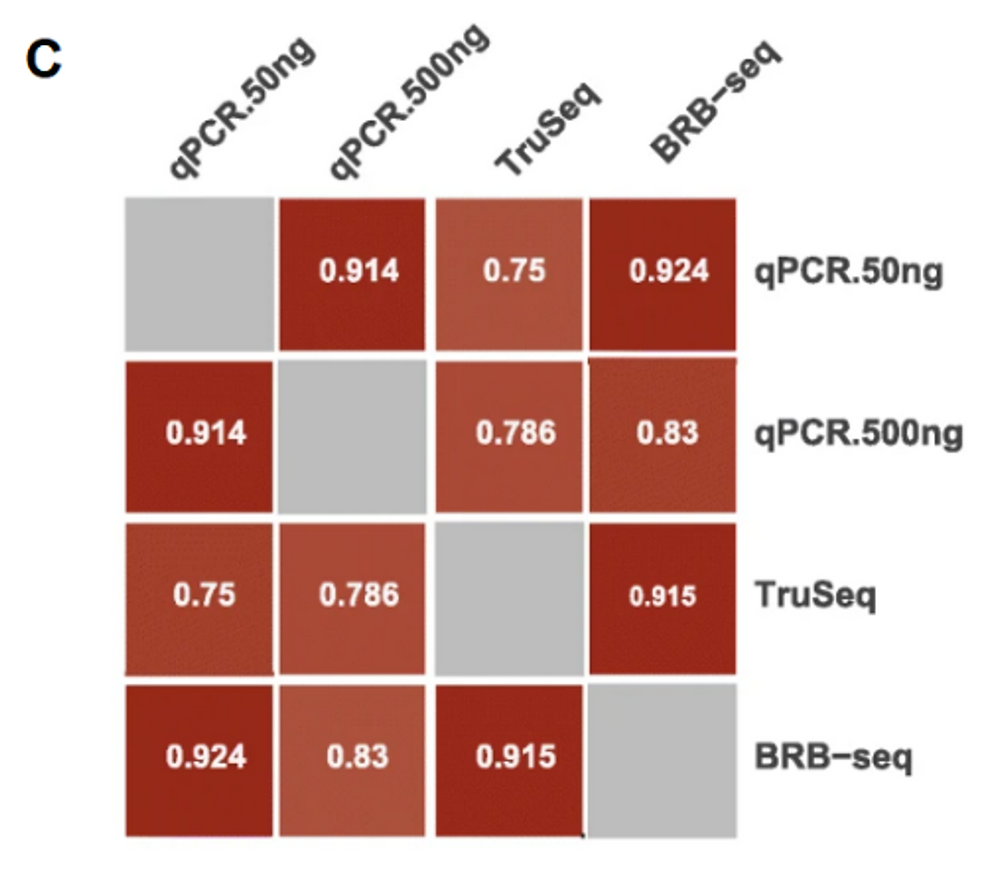
Finally, a direct comparison of BRB-seq with qPCR shows an even greater correlation with qPCR than TruSeq (Fig. C), especially with low RNA quantities, which again goes in the direction of BRB-seq being a good alternative to qPCR.
In conclusion, we anticipate that BRB-seq will become an attractive alternative for routine gene expression analysis and ultimately could replace large RT-qPCR assays. Indeed, the evaluation of the expression of a few qPCR reactions (a few genes in triplicate) will cost approximately the same or even more than one full transcriptome analysis produced by BRB-seq, which involves library preparation and sequencing expenses. Importantly, low library preparation cost and time imply that more replicates can be profiled, which will greatly increase the statistical power underlying any differential expression analysis. The only limitation we envision would be the necessity of a platform that can be used by experimental biologists to handle BRB-seq data and produce ready-to-use count matrices in a straightforward manner, therefore further streamlining the BRB-seq transcriptomics to the extent of a mere qPCR experiment.