
With a growing number of published 3’ mRNA-seq methods now available, researchers have more choices than ever for high-throughput and cost-effective transcriptomic screening. While broadly similar, 3’ mRNA-seq protocols often differ in how they barcode and multiplex samples, synthesize cDNA, and prepare libraries, which can impact cost, throughput, and data quality.
Whether you’re screening drug candidates, studying gene function, or profiling toxicology responses, your choice of transcriptomics method matters.
So, let’s dig deep into how MERCURIUS™ BRB-seq and MERCURIUS™ DRUG-seq differ from other published high-throughput 3’ mRNA-seq methods, like HiCAR-seq, Prime-seq, and BOLT-seq, and how they perform compared to standard RNA-seq approaches.
What are the differences in 3’ mRNA-seq workflows?
While the workflow of each published method we discuss is broadly similar, there are some key differences in how they achieve core processes like sample barcoding and reverse transcription (Fig. 1). These differences ultimately influence hands-on time, cost, and overall performance.
For instance, BOLT-seq omits certain stages common to other workflows to decrease hands-on time, but with the trade-off of potential impacts on performance compared to standard RNA-seq methods (Choi et al., 2024).
See our companion article for the features of different published 3’ mRNA-seq methods.
In this article, we’ve focused on key stages. However, there are also differences in the type and number of washing and purification processes throughout each protocol.
You can find the complete protocol for Alithea’s MERCURIUS™ DRUG-seq here and MERCURIUS™ BRB-seq here.
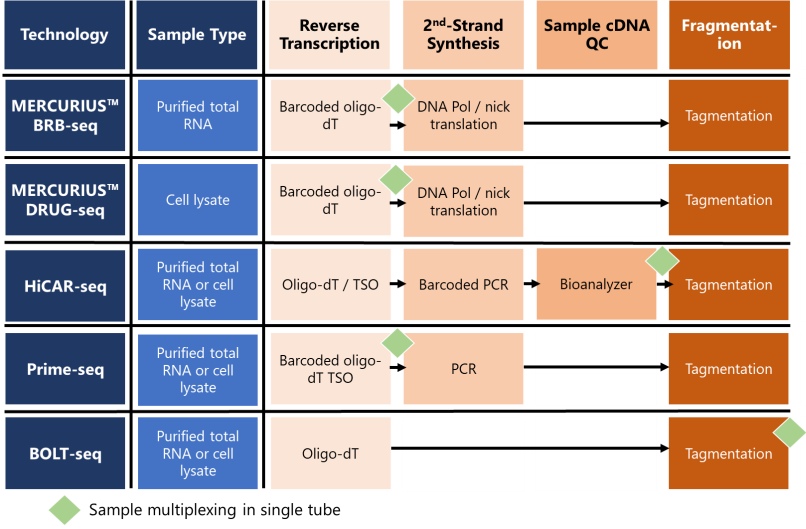
Figure 1. Schematic table highlighting the differences in key stages of published 3’ mRNA-seq workflows.
What starting material can you use for 3’ mRNA-seq?
3’ mRNA-seq technologies are optimized for different starting samples, like purified RNA or cell lysates. MERCURIUS™ BRB-seq is the only method we discuss that is solely optimized for purified RNA from a wide range of sample types, including cells, blood, and organoids (Fig. 1).
In contrast, MERCURIUS™ DRUG-seq is optimized only for cell lysates, whereas HiCAR-seq, Prime-seq, and BOLT-seq work with both purified total RNA and cell lysates.
Preparing libraries directly on cell lysates avoids time-consuming and costly RNA extraction and purification stages, which makes RNA-extraction-free approaches, like MERCURIUS™ DRUG-seq, suited to automation in drug discovery or toxicology screening pipelines.
When and how are samples barcoded and multiplexed?
Different 3’ mRNA-seq methods employ different approaches for sample barcoding at varying workflow stages. Crucially, this influences when samples are pooled, alongside other technical considerations.
In MERCURIUS™ BRB-seq and MERCURIUS™ DRUG-seq, highly optimized barcoded oligo-dT primers uniquely tag the 3’ poly(A)-tail of mRNA molecules in each RNA sample during the reverse transcription step of cDNA library preparation (Fig. 1). Sample pooling into the same tube follows.
The rigorously verified MERCURIUS™ oligos include an adaptor for primer annealing, a specific sample barcode for each RNA sample, and a unique molecular identifier (UMI) that tags each mRNA molecule with a unique nucleotide sequence to detect mRNA duplicates that result from PCR amplification.
Prime-seq barcodes also include UMIs and are added at the reverse transcription, followed by pooling of all samples, whereas HiCAR-seq adds barcoded UMI PCR primers at the second-strand synthesis step for mid-stage sample pooling (Janjic et al., 2022; Veeranagouda et al., 2020). BOLT-seq barcodes samples at the final stage of cDNA library amplification, but doesn’t include UMIs as standard. This is followed by late-stage pooling of completed individual cDNA libraries at the end of the workflow (Choi et al., 2024) (Fig. 1).
To template switch or not?
While the MERCURIUS™ approaches and Prime-seq both use barcoded oligo-dT primers to uniquely tag mRNA samples at the reverse transcription stage, one crucial difference is that first-strand synthesis is performed with reverse transcriptase and a template switching oligo (TSO) for Prime-seq but with DNA polymerase I nick translation for the MERCURIUS™ technologies (Fig. 1).
Template switching via a TSO allows the generation of full-length cDNAs during first-strand synthesis. Upon reaching the 5′ end of the mRNA template, the terminal transferase activity of particular reverse transcriptases adds additional deoxycytidine nucleotides to the 3′ end of the newly synthesized cDNA strand. These bases act as a TSO hybridization site that allows the reverse transcriptase to “switch” template strands and continue replication to the 5′ end. This generates cDNAs containing the complete transcript and barcode sequences of choice.
However, template switching can cause artifacts and unsystematic biases in sequencing data via a process called strand invasion, which interrupts first-strand synthesis and results in cDNAs that are artificially shorter than the mRNA template due to the incomplete reverse transcription (Tang et al., 2013).
Crucially, MERCURIUS™ approaches don’t require a TSO during the first-strand synthesis, thanks to the use of the ‘Gubler-Hoffman method’, which drives second-strand generation by RNA primer-dependent nick translation with DNA polymerase I (Gubler and Hoffman, 1983). This alleviates the associated bias and potential artifacts related to strand invasion observed with TSO (Alpern et al., 2019).
Regardless of how it’s achieved, barcoding early in the workflow with both MERCURIUS™ and Prime-seq technologies allows sample multiplexing just after reverse transcription, for subsequent processing of up to 384 samples for MERCURIUS™ methods and 96 samples for Prime-seq in a single tube.
How is reverse transcription performed?
The method of priming reverse transcription is similar for all methods and relies on oligo-dT primers that all target the poly-A tail of mRNA molecules (Fig. 1). The reverse transcriptase used differs for each method.
HiCAR-seq uses SMARTScribe RT with TSO, Prime-seq uses Maxima H Minus enzyme with TSO, BOLT-seq uses in-house purified M-MuLV to reduce costs, whereas the published BRB-seq study found little difference between Maxima H Minus and Superscript II reverse transcriptase (Alpern et al., 2019).
The published DRUG-seq protocol used Maxima RT with a TSO (Ye et al., 2018). However, the commercial MERCURIUS™ technologies are now optimized with a high-fidelity enzyme, avoiding the use of TSO for more robust reverse transcription and removal of TSO-associated biases.
And second-strand synthesis?
Following first-strand cDNA synthesis, second-strand synthesis then occurs. For MERCURIUS™ technologies, DNA polymerase I nick translation eliminates the need for TSO required in PCR-based second-strand synthesis, like HiCAR-seq and Prime-seq methods (Fig. 1).
In contrast, BOLT-seq completely omits second-strand synthesis and pre-amplification by directly subjecting RNA/DNA hybrid duplexes from first-strand synthesis to Tn5 transposase-mediated tagmentation (Fig. 1).
Can you QC cDNA from individual samples?
All of the 3’ mRNA-seq protocols we discuss are designed to be rapid, high-throughput, and low-cost, so they usually omit any per-sample quality control (QC) during the workflow. To ensure balanced read distribution across all samples after sequencing, researchers should start with similar cell numbers or RNA amounts, which are then pooled in equimolar ratios for MERCURIUS™ technologies, Prime-seq, and BOLT-seq.
As no per-sample QC steps are included in these methods, any variations in cellular or RNA inputs across different wells risk over-representation in sequencing runs, which are only detected upon data analysis. However, MERCURIUS™ technologies allow cost-effective shallow sequencing to ensure libraries are of sufficient quality before deeper sequencing if sample quality is a concern.
The only protocol that facilitates within-workflow QC is HiCAR-seq, which allows Bioanalyzer assessment of individual cDNA samples before pooling, but this adds additional time and cost to the workflow.
How are libraries fragmented and indexed?
All workflows fragment and index libraries with tagmentation using in-house Tn5 transposase or use the Nextera XT Kit. The original BRB-seq publication showed minimal impact of using an in-house Tn5 variant or the Nextera Tn5 enzyme, as they had relatively minor effects on sequencing data (Alpern et al., 2019).
How well do 3’ mRNA-seq methods perform?
With any novel method, it’s crucial to benchmark its performance against established technologies to ensure it provides robust, reliable, and reproducible data suitable for testing diverse hypotheses.
For most of the published 3’ mRNA-seq methods we’ve discussed, researchers compared the results to traditional full-length bulk RNA-seq technologies to evaluate how many genes were detected, the number of differentially expressed genes, and other technical metrics.
1. BRB-seq
In the initial BRB-seq paper, Alpern et al. showed that BRB-seq detected a comparable number of expressed and differentially expressed genes as the Illumina TruSeq Stranded mRNA protocol at the same sequencing depths per sample (Fig. 1A). Log2 read counts were highly correlated between BRB-seq technical replicates (Pearson’s correlation r = 0.987) and TruSeq libraries (Pearson’s correlation r = 0.920) (Fig. 1B). This benchmarking highlighted BRB-seq as a robust but more cost-effective and scalable alternative to traditional RNA-seq.
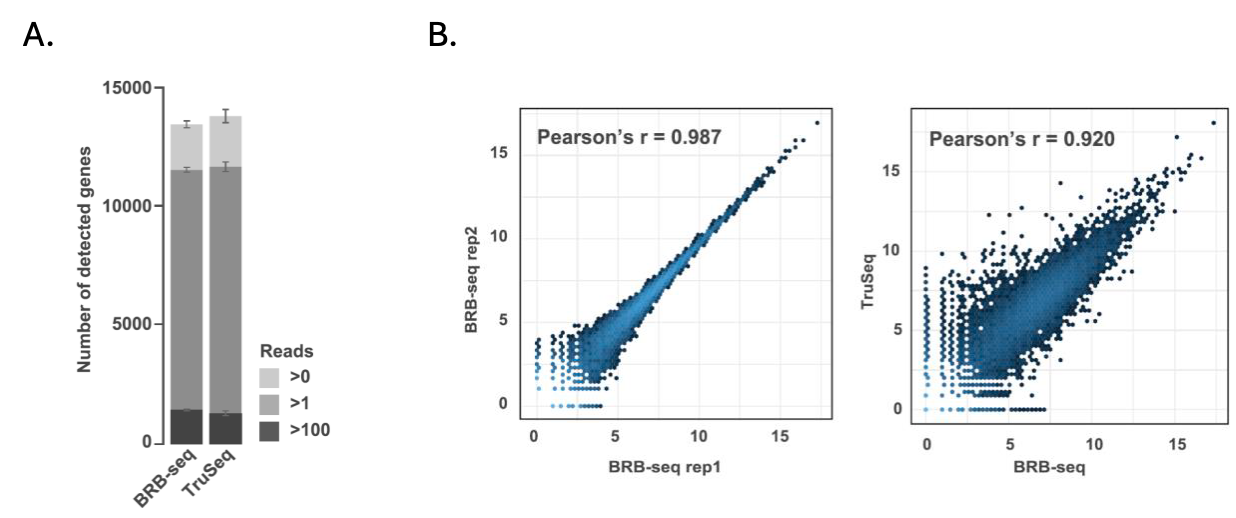
Figure 1. Benchmarking BRB-seq against TruSeq data. Modified from Alpern et al., 2019.
2. DRUG-seq
The authors of the initial DRUG-seq papers also benchmarked the technology against standard RNA-seq, although not at similar sequencing depths. They found that DRUG-seq detected a median of 12,000 genes at 13 million reads/well and 11,000 genes at two million reads/well, compared to a median of 17,000 expressed genes detected in standard RNA-seq at 42 million reads/well (Fig. 2) (Ye et al., 2018). Genes detected as differentially expressed by DRUG-seq were also broadly consistent with those identified by standard RNA-seq.
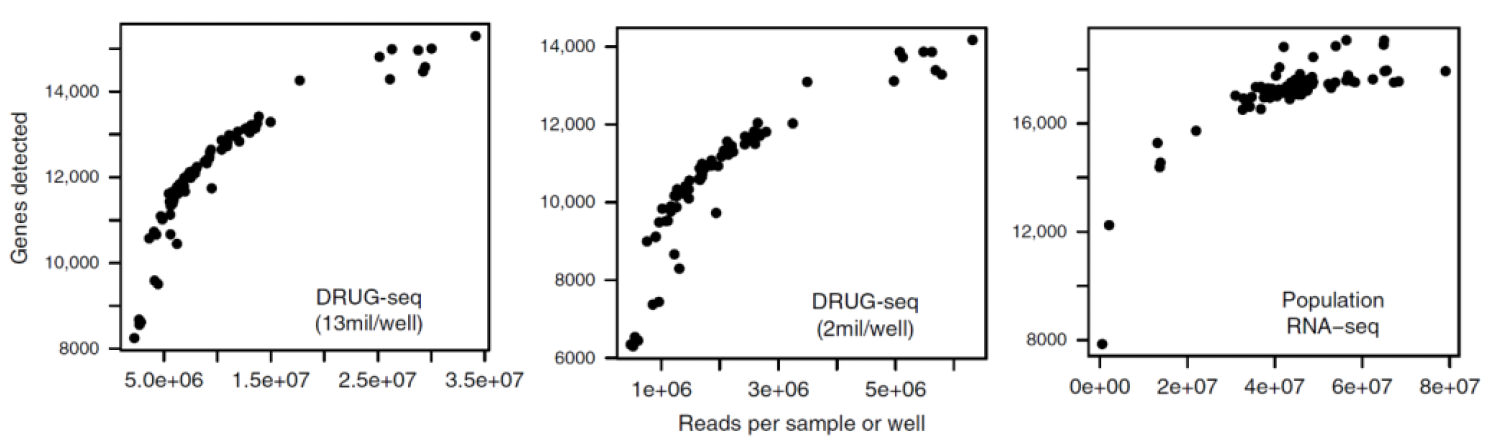
Figure 2. Benchmarking DRUG-seq against population-level TruSeq data. Modified from Ye et al., 2018.
Commercialized MERCURIUS™ DRUG-seq has improved the published DRUG-seq protocol by avoiding bias-prone template switching and pre-amplification stages for a more sensitive and streamlined protocol that detects similar numbers of genes as BRB-seq (Fig. 3A) and NEBNext (Fig. 3B).
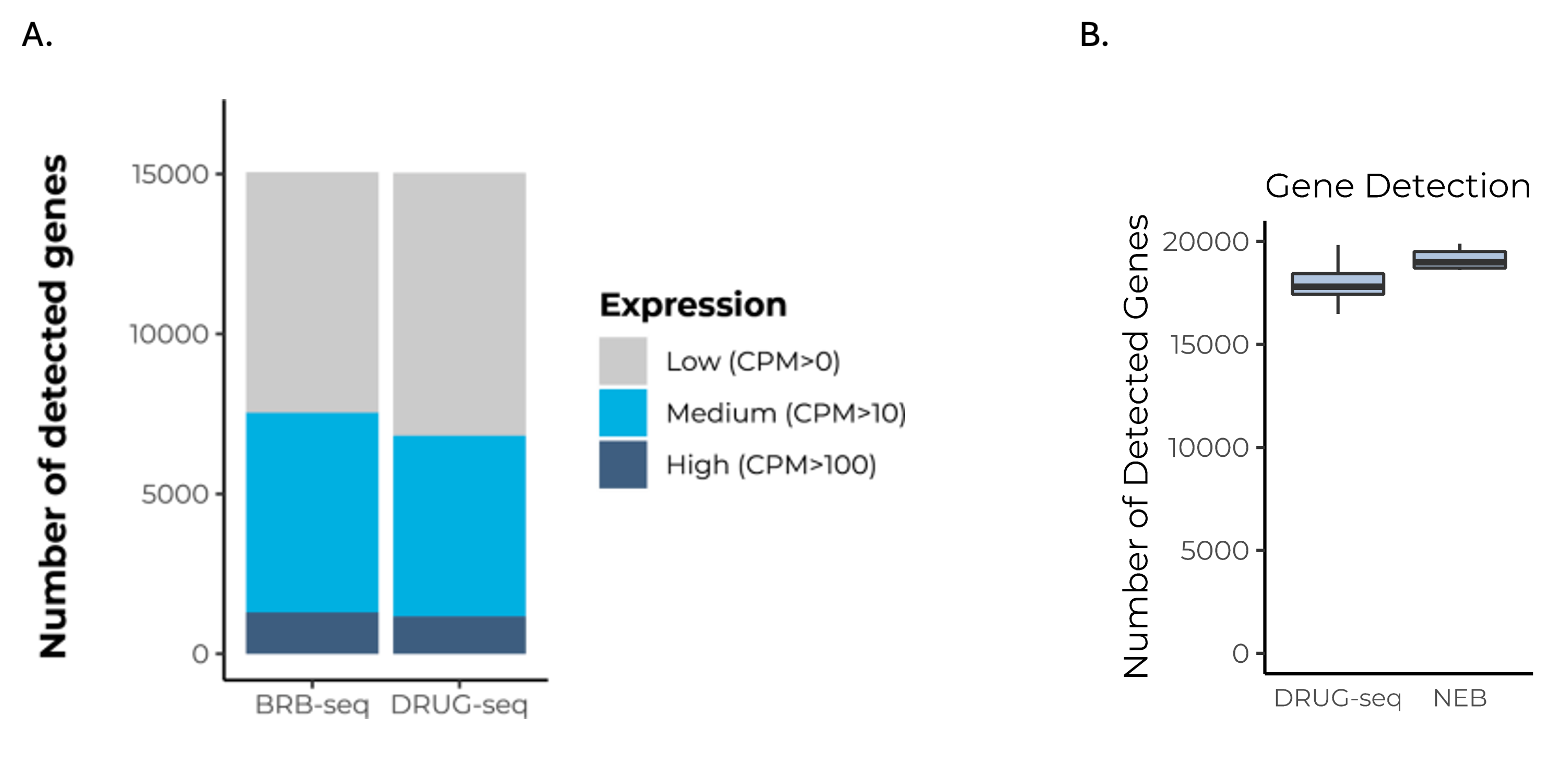
Figure 3. MERCURIUS™ DRUG-seq detects as many genes as MERCURIUS™ BRB-seq and NEBNext, but is RNA-extraction-free.
3. Prime-seq
Prime-seq also performs similarly to Illumina TruSeq, as comparable numbers of expressed genes were detected at the same sequencing depths (Fig. 4) (Janjic et al., 2022). Prime-seq and TruSeq libraries were also highly correlated between observed expression levels and known concentrations of control RNA (ERCC) spike-ins (Pearson’s correlation r = 0.94), indicating method accuracy.
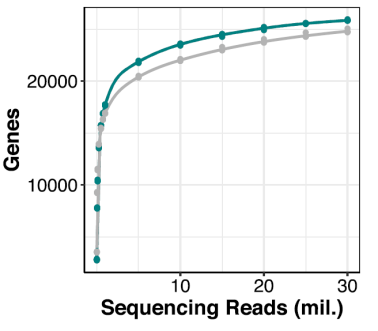
Figure 4. Benchmarking the number of genes detected with Prime-seq (green) against TruSeq (grey) at different sequencing depths. Modified from Janjic et al., 2022.
4. HiCAR-seq performance
The authors of the initial HiCAR-seq paper provided no systematic, side-by-side comparison with conventional RNA-seq in terms of transcript quantification accuracy or differential expression analysis (Veeranagouda et al., 2020).
5. BOLT-seq performance
In the initial manuscript, BOLT-seq was compared to the standard RNA-seq NEBNext method. BOLT-seq detected only 1185 differentially expressed genes compared to 3296 for NEBNext, suggesting a loss of detection capacity compared to full-length RNA-seq methods (Fig. 5) (Choi et al., 2024). Despite this, for the detected differentially expressed genes, the correlation between the log2-fold change of DE genes detected by NEBNext and BOLT-seq was high (Pearson’s correlation r = 0.944).
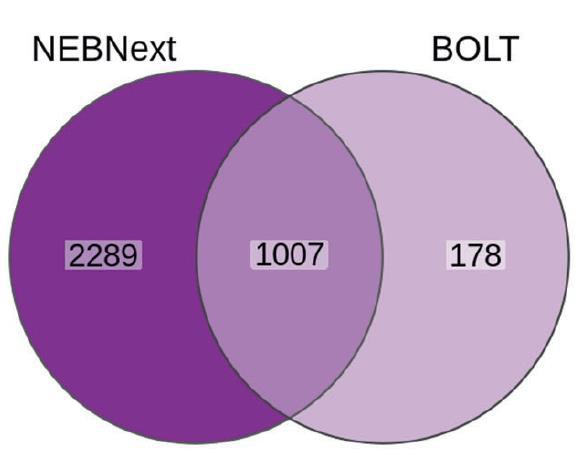
Figure 5. Comparison of differentially expressed genes detected by NEBNext and BOLT-seq. Modified from Choi et al., 2024.
3’ mRNA-seq is robust and reliable for large-scale transcriptomics
Overall, published bulk 3’ mRNA-seq methods are revolutionizing transcriptomic screens with unprecedented sample throughput, cost-efficiency, and comparable performance to gold standard RNA-seq methods.
Commercial methods like MERCURIUS™ DRUG-seq and MERCURIUS™ BRB-seq combine the convenience of “buy and go” kits or services with ultra-cost-effective and accurate workflows necessary for reproducible, highly informative screening pipelines that might uncover your next lead, mechanism of action, or off-target effect.
Ready to accelerate your next screen? Explore our MERCURIUS™ kits or speak with our team to find the best 3′ mRNA-seq solution for your project.
References
- Alpern, D., Gardeux, V., Russeil, J., Mangeat, B., Meireles-Filho, A.C., Breysse, R., Hacker, D. and Deplancke, B., 2019. BRB-seq: ultra-affordable high-throughput transcriptomics enabled by bulk RNA barcoding and sequencing. Genome biology, 20, pp.1-15.
- Choi, J., Hyun, J., Hyun, J., Kim, J.H., Lee, J.H. and Bang, D., 2024. Cost and time-efficient construction of a 3′-end mRNA library from unpurified bulk RNA in a single tube. Experimental & Molecular Medicine, 56(2), pp.453-460.
- Gubler U, Hoffman BJ. A simple and very efficient method for generating cDNA libraries. Gene. 1983;25:263–9.
- Janjic, A., Wange, L.E., Bagnoli, J.W., Geuder, J., Nguyen, P., Richter, D., Vieth, B., Vick, B., Jeremias, I., Ziegenhain, C. and Hellmann, I., 2022. Prime-seq, efficient and powerful bulk RNA sequencing. Genome biology, 23(1), p.88.
- Tang, D.T., Plessy, C., Salimullah, M., Suzuki, A.M., Calligaris, R., Gustincich, S. and Carninci, P., 2013. Suppression of artifacts and barcode bias in high-throughput transcriptome analyses utilizing template switching. Nucleic acids research, 41(3), pp.e44-e44.
- Veeranagouda, Y., Zachayus, J.L., Guillemot, J.C., Venier, O. and Didier, M., 2020. High-Throughput Cellular RNA Sequencing (HiCAR-Seq): Cost-Effective, High-Throughput 3′ mRNA-Seq Method Enabling Individual Sample Quality Control. Current Protocols in Molecular Biology, 132(1), p.e123.
- Ye, C., Ho, D.J., Neri, M., Yang, C., Kulkarni, T., Randhawa, R., Henault, M., Mostacci, N., Farmer, P., Renner, S. and Ihry, R., 2018. DRUG-seq for miniaturized high-throughput transcriptome profiling in drug discovery. Nature communications, 9(1), p.4307.