
3’ mRNA-seq technologies are gradually revolutionizing high-throughput bulk transcriptomics.
Large-scale toxicology, drug discovery, or genetic perturbation screens often assess thousands of experimental conditions, compounds, or dosages in parallel to generate information-rich data crucial for risk-aware decision-making in pipelines.
However, traditional transcriptomic methods like bulk RNA-seq are slow and expensive due to overly involved workflows, limited sample multiplexing possibilities, and the increased sequencing depth required for robust transcriptome-wide data.
Fortunately, continual innovation has led to the publication and commercialization of numerous massively multiplexed 3’ mRNA-seq methods that now provide researchers with the tools to generate cost-effective, transcriptome-wide data at scale.
Here, we provide an overview of 3’ mRNA-seq, highlight the similarities and differences of some recently published methods, and indicate why opting for commercial over ‘home-brew’ options might be better in the long run.
What is 3’ mRNA-seq?
Different flavors of published or commercialized 3’ mRNA-seq technologies exist, but their overall principle is broadly similar. 3’ mRNA-seq refers to quantitative, transcriptome-wide library preparation technologies that use unique molecular barcodes to tag the 3’ end of polyadenylated RNA molecules. Different technologies are optimized for purified RNA (see MERCURIUS™ BRB-seq) or are designed to work directly on crude cell lysates and completely omit time-consuming and costly RNA-extraction stages (see MERCURIUS™ DRUG-seq).
Unlike standard bulk RNA-seq, which sequences full-length transcripts, 3′ mRNA-seq focuses on preparing only 3 ′ fragments of tagged mRNA molecules for sequencing. While information about isoforms and splice variants is lost, this information isn’t required for most studies performing differential expression analyses of protein-coding genes between experimental conditions. Thus, the 3’ barcoded nature of the technology means that sequencing space is used more efficiently to multiplex more samples for higher-throughput studies at a lower cost. A focus on capturing only the 3’ region also means it excels at assessing highly degraded RNA common in FFPE samples, while providing broader, unbiased coverage than other high-throughput gene expression profiling options like probe-based, targeted profiling technologies.
Far fewer reads are required to robustly quantify transcriptome-wide gene expression than full-length RNA-seq methods (~2 to 5 million vs. ~30 million reads per sample), without sacrificing gene detection or data quality (Alpern et al., 2019; Ma et al., 2019). Overall, the approach maximizes the amount of expression data generated while dramatically reducing cost. This makes it suitable for many exploratory expression analyses, including uncovering drug mechanisms of action, toxicogenomic effects, or on-/off-targets of compounds.
Learn more about 3’ mRNA-seq.
Comparison of published 3’ mRNA-seq methods
With the growing number of published 3’ mRNA-seq methods, we briefly summarize the main features of different approaches (Table 1). We previously covered how MERCURIUS™ BRB-seq compares to two commercially available methods here, so we focus on recently published methods, including MERCURIUS™ BRB-seq, MERCURUS™ DRUG-seq, HiCAR-seq, Prime-seq, and BOLT-seq. See our accompanying blog post for detailed comparisons of the different workflows.
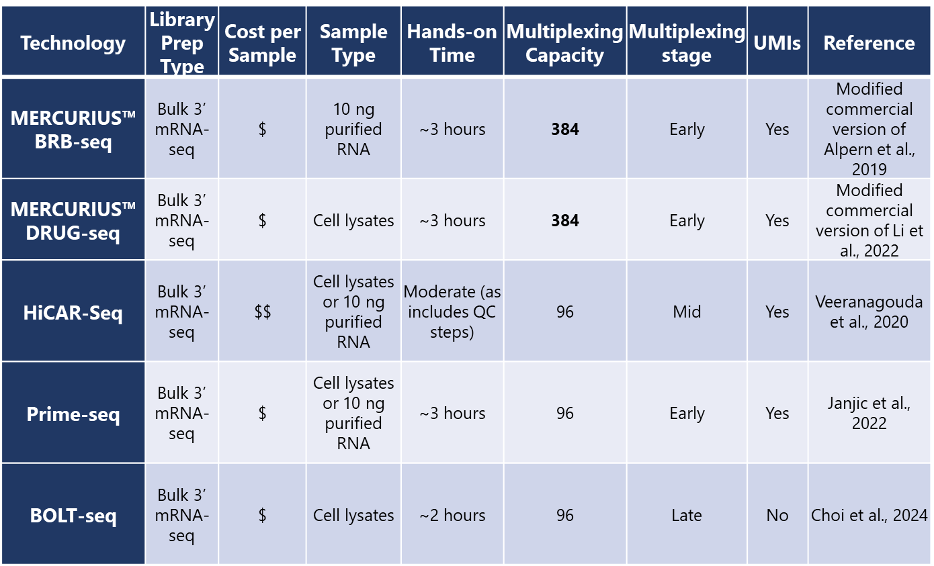
Table 1. Core features of different published 3’ mRNA-seq methods
Key considerations when selecting a method
1. Cost per sample
For most high-throughput studies, cost is a major consideration. Costs can spiral if overly expensive methods are used to profile thousands of samples. All of the 3’ mRNA-seq technologies in Table 1 are significantly cheaper than traditional RNA-seq methods.
For instance, Prime-seq is fourfold more cost-effective than Illumina TruSeq. MERCURIUS™ BRB-seq and DRUG-seq are also among the most data-rich but cost-effective methods available. Other methods have similar costs, apart from HiCAR-seq, which includes a more expensive Bioanalyzer intermediate QC for more precise pooling of equal concentrations of cDNA from different samples.
2. Sample type
Some 3’ mRNA-seq methods are optimized either for purified RNA samples or for cell lysates as input, but some methods offer the flexibility for both sample types. For large-scale experiments with automation in mind, streamlined processes are a necessity, so protocols with integrated cell lysis that are completely RNA-extraction-free could be the optimal choice. These technologies only require per-well drug treatments, followed by direct cell lysis and library preparation. Ultimately, the most suitable method depends on sample type and research question.
3. Hands-on time
More samples also mean potentially more time to results. All the methods discussed have overall workflows of approximately one day, with hands-on times ranging from two to three hours. The speed of these approaches allows you to reduce the time from sample to results compared to standard RNA-seq methods.
BOLT-seq is the quickest method as it completely omits second-strand cDNA synthesis and pre-amplification by directly subjecting RNA/DNA hybrid duplexes to Tn5 transposase-mediated tagmentation.
4. Sample Multiplexing
Multiplexing as many samples as possible with sample barcodes, without compromising data quality, is one of the most important considerations for any high-throughput screening experiment. MERCURIUS™ BRB-seq and MERCURIUS™ DRUG-seq offer the highest level of sample multiplexing in a single tube of any other published technology we’ve covered.
By assessing up to 384 samples instead of only 96 samples like other methods, consumable usage per sample is dramatically reduced with MERCURIUS™ technologies, further lowering costs while ensuring sustainability.
5. Multiplexing Stage
Traditional RNA-seq workflows often multiplex samples just before sequencing, meaning technical errors and sample loss can creep in when treating each sample individually. MERCURIUS™ BRB-seq, MERCURIUS™ DRUG-seq, and Prime-seq employ early multiplexing around the reverse transcription stage, when all samples are pooled into one tube. This reduces consumable usage, hands-on time, likelihood of technical errors, and cost. In contrast, HiCAR-seq multiplexes samples midway through the workflow after per-sample quality control, and BOLT-seq multiplexes completed sample libraries just before sequencing.
6. Unique molecular identifier inclusion
Unique molecular identifiers (UMIs) tag each mRNA molecule to allow users to correct for PCR duplicate reads during data analysis. They ensure more robust results by removing reads that result from technical artifacts, instead of true biological effects. BOLT-seq is the only technology we’ve included that doesn’t include UMIs as standard.
Benefits of commercial 3’ mRNA-seq technologies
When preparing libraries for bulk 3′ mRNA sequencing, researchers can choose between commercial kits like MERCURIUS™ BRB-seq and MERCURIUS™ DRUG-seq or published non-commercialized protocols. Commercial kits offer convenience, consistency, and on-demand technical support, making them ideal for labs seeking standardized workflows with minimal optimization. This is especially important in regulated settings or multi-lab collaborative studies where reagents might vary considerably for DIY options.
MERCURIUS™ BRB-seq and MERCURIUS™ DRUG-seq are also offered as a service to further streamline workflows. Users send 96- or 384-well plates with frozen cells to our service centers (located in Switzerland and the United States), and the Alithea Genomics team processes the plates and returns results in as little as two weeks.
Overall, commercial kits are well-validated, rigorously optimized, and highly compatible with automated platforms, and their speed and ease of use are often acceptable trade-offs for any limitations in protocol flexibility compared to in-house protocols. DIY protocols also demand more technical expertise, rigorous quality control, and time investment to ensure reproducibility than ‘buy and go’ commercial options. Ultimately, the decision depends on the specific goals, budget, and technical capacity of the lab.
Ready to accelerate your next screen? Explore our MERCURIUS™ kits or speak with our team to find the best 3′ mRNA-seq solution for your project.
References
- Alpern, D., Gardeux, V., Russeil, J., Mangeat, B., Meireles-Filho, A.C., Breysse, R., Hacker, D. and Deplancke, B., 2019. BRB-seq: ultra-affordable high-throughput transcriptomics enabled by bulk RNA barcoding and sequencing. Genome biology, 20, pp.1-15.
- Choi, J., Hyun, J., Hyun, J., Kim, J.H., Lee, J.H. and Bang, D., 2024. Cost and time-efficient construction of a 3′-end mRNA library from unpurified bulk RNA in a single tube. Experimental & Molecular Medicine, 56(2), pp.453-460.
- Janjic, A., Wange, L.E., Bagnoli, J.W., Geuder, J., Nguyen, P., Richter, D., Vieth, B., Vick, B., Jeremias, I., Ziegenhain, C. and Hellmann, I., 2022. Prime-seq, efficient and powerful bulk RNA sequencing. Genome biology, 23(1), p.88.
- Li, J., Ho, D.J., Henault, M., Yang, C., Neri, M., Ge, R., Renner, S., Mansur, L., Lindeman, A., Kelly, B. and Tumkaya, T., 2022. DRUG-seq provides unbiased biological activity readouts for neuroscience drug discovery. ACS Chemical Biology, 17(6), pp.1401-1414.
- Veeranagouda, Y., Zachayus, J.L., Guillemot, J.C., Venier, O. and Didier, M., 2020. High‐Throughput Cellular RNA Sequencing (HiCAR‐Seq): Cost‐Effective, High‐Throughput 3′ mRNA‐Seq Method Enabling Individual Sample Quality Control. Current Protocols in Molecular Biology, 132(1), p.e123.